Internet Book of Critical Care (IBCC)
Online Medical Education on Emergency Department (ED) Critical Care, Trauma, and Resuscitation


Pulmonary hypertension (PH)
February 10, 2024 by Josh Farkas
- Functional assessment
- Chest radiograph
- Echocardiography
- Vasoreactivity testing
- Evaluation for PH of unknown etiology
- Basic supportive care for PH
- Calcium channel blockers
- Endothelin-receptor antagonists
- Phosphodiesterase-5 inhibitors & guanylate cyclase stimulators
- Systemic prostacyclins
- Oral prostacyclin receptor agonist (selexipag)
- Inhaled prostacyclin analogues
- Scleroderma-related PH ➡️
- PAH associated with HIV
- PAH associated with portal hypertension
- PAH associated with congenital heart disease
- Pulmonary veno-occlusive disease/pulmonary capillary hemangiomatosis
- Group 2 PH (left heart failure)
- Group 3 PH (lung disease)
- Group 4 PH (chronic thromboembolic pulmonary hypertension)
- PH in sickle cell disease
- PH in sarcoidosis ➡️
- Questions & discussion
abbreviations used in the pulmonary section: 9
- ABPA : Allergic bronchopulmonary aspergillosis 📖
- AE-ILD : Acute exacerbation of ILD 📖
- AEP : Acute eosinophilic pneumonia 📖
- AFB : Acid-fast bacilli
- AIP : Acute interstitial pneumonia (Hamman-Rich syndrome) 📖
- ANA : Antinuclear antibody 📖
- ANCA : Antineutrophil cytoplasmic antibodies 📖
- ARDS : Acute respiratory distress syndrome 📖
- ASS : Antisynthetase syndrome 📖
- BAL : Bronchoalveolar lavage 📖
- BiPAP : Bilevel positive airway pressure 📖
- CEP : Chronic eosinophilic pneumonia 📖
- CF : Cystic fibrosis 📖
- COP : Cryptogenic organizing pneumonia 📖
- CPAP : Continuous positive airway pressure 📖
- CPFE : Combined pulmonary fibrosis and emphysema 📖
- CTD-ILD : Connective tissue disease-associated interstitial lung disease 📖
- CTEPH : Chronic thromboembolic pulmonary hypertension 📖
- DAD : Diffuse alveolar damage 📖
- DAH : Diffuse alveolar hemorrhage 📖
- DIP : Desquamative interstitial pneumonia 📖
- DLCO : Diffusing capacity for carbon monoxide 📖
- DRESS : Drug reaction with eosinophilia and systemic symptoms 📖
- EGPA : Eosinophilic granulomatosis with polyangiitis 📖
- FEV1 : Forced expiratory volume in 1 second 📖
- FVC : Forced vital capacity 📖
- GGO : Ground-glass opacity 📖
- GLILD : Granulomatous and lymphocytic interstitial lung disease 📖
- HFNC : High flow nasal cannula 📖
- HP : Hypersensitivity pneumonitis 📖
- IPAF : Interstitial pneumonia with autoimmune features 📖
- IPF : Idiopathic pulmonary fibrosis 📖
- IVIG : Intravenous immunoglobulin 📖
- LAM : Lymphangioleiomyomatosis 📖
- LIP : Lymphocytic interstitial pneumonia 📖
- MAC : Mycobacterium avium complex 📖
- MCTD : Mixed connective tissue disease 📖
- NIV : Noninvasive ventilation (including CPAP or BiPAP) 📖
- NSIP : Nonspecific interstitial pneumonia 📖
- NTM : Non-tuberculous mycobacteria 📖
- OHS : Obesity hypoventilation syndrome 📖
- OP : Organizing pneumonia 📖
- OSA : Obstructive sleep apnea 📖
- PAP : Pulmonary alveolar proteinosis 📖
- PE : Pulmonary embolism 📖
- PFT : Pulmonary function test 📖
- PLCH : Pulmonary Langerhans cell histiocytosis 📖
- PPFE : Pleuroparenchymal fibroelastosis 📖
- PPF : Progressive pulmonary fibrosis 📖
- PVOD/PCH Pulmonary veno-occlusive disease/pulmonary capillary hemangiomatosis 📖
- RB-ILD : Respiratory bronchiolitis-associated interstitial lung disease 📖
- RP-ILD : Rapidly progressive interstitial lung disease 📖
- TNF : Tumor necrosis factor
- UIP : Usual interstitial pneumonia 📖
(back to contents)
(#1) initial symptoms
- Onset is typically insidious, often leading to delayed diagnosis.
- Dyspnea on exertion is the most frequent presentation.
(#2) right ventricular congestive symptoms
- Right ventricular failure causes peripheral edema and weight gain.
- Bowel edema may cause malnutrition, anorexia/nausea.
- Hepatic congestion may cause abdominal fullness, discomfort.
- Ascites can occur.
(#3) impaired cardiac output (most worrisome)
- Exertional syncope or presyncope.
- Cool extremities.
- Prolonged capillary refill.
- Hypotension and tachycardia can be seen.
other symptoms:
- Palpitations.
- Anginal chest pain (due to subendocardial ischemia of the right ventricle, or dynamic compression of the left main coronary artery).
- Hoarseness due to Ortner syndrome : dilated pulmonary artery impairs the left recurrent laryngeal nerve, causing left vocal cord paralysis. Recurrent laryngeal nerve dysfunction may also result from dilation of the left atrium or aorta (cardiovocal syndrome). ( 31174660 )
- Hemoptysis (rare, may be severe).
The NYHA functional assessment and the World Health Organization (WHO) functional status are essentially the same:
- Class I = Asymptomatic .
- Mild limitation, only with activity.
- Ordinary physical activity causes symptoms (abnormal dyspnea/fatigue, chest pain, or near syncope).
- Marked limitation with activity.
- Comfortable at rest, without rest symptoms.
- Any activity causes symptoms.
- May have symptoms at rest.
- Signs of right ventricular failure.

dilated right atrium and right ventricle
- Frontal radiograph: Prominent bulge of the right atrium, to the right side of the spine.
- Lateral radiograph: Dilation of the right ventricle causes a reduction in the retrosternal air space. The right ventricle may contact >1/3 of the sternum.
enlarged central pulmonary arteries
- The convexity of the main pulmonary artery may be prominent.
- Prominent pulmonary arteries may mimic hilar lymphadenopathy. However, if arteries can be seen converging on the enlarged pulmonary artery, this confirms the presence of pulmonary artery enlargement (“hilum convergence sign”).
- Arteries may rapidly taper, with peripheral oligemia.
(#1) pulmonary artery diameter enlargement
- This ratio has a fairly high specificity for identifying pulmonary hypertension (~90%). (Walker 2019)
- The ratio is inaccurate among patients with aortic dilation (more likely in older patients).
- Specificity may be enhanced by using a cutoff of >32.5 mm. (Walker 2019)

(#2) chamber dilation and venous congestion
- Enlarged right heart chambers (right atrium, right ventricle).
- Inferior vena cava dilation.
- Contrast reflux into the inferior vena cava and sometimes even hepatic veins.
other findings may include:
- If 3 of the 4 segmental arteries are enlarged and the main pulmonary artery is >29 mm, this is highly specific for pulmonary hypertension. (Fishman 2023)
- This ratio may not be accurate in the presence of pulmonary pathology that affects bronchus caliber (e.g., bronchiectasis).
- RV outflow tract wall hypertrophy (>6 mm). ( 36017548 )
- Pericardial effusion may be seen in some cases.
- Longstanding pulmonary hypertension may cause cholesterol granulomas to occur within alveolar spaces.
- Radiologically these may appear as diffuse centrilobular GGO (ground-glass opacities). ( 24791617 ) The differential diagnosis of this finding includes PCH (pulmonary capillary hemangiomatosis).
signs of the cause of pulmonary hypertension
- Parenchymal lung abnormalities suggest the possibility of Group III pulmonary hypertension.
- Dilation of the left atrium suggests the presence of left heart disease (i.e., Group II pulmonary hypertension).
- Pleural effusion is generally not caused by pulmonary hypertension, so this suggests the presence of another process (e.g., left heart disease or primary lung disease). (Murray 2022)
- May be seen in severe idiopathic PAH (pulmonary atrial hypertension) due to repeated microhemorrhages.
- May occur in pulmonary capillary hemangiomatosis. 📖
- PCH/PVD (pulmonary capillary hemangiomatosis/pulmonary veno-occlusive disease) may cause features that resemble cardiogenic pulmonary edema. 📖
qualitative signs of pulmonary hypertension may be seen
- Dilated coronary sinus (although this may also be incidental, due to a persistent left superior vena cava, or due to congenital heart disease).
- Right atrial dilation.
- Right ventricular dilation (e.g., basal RV diameter greater than LV diameter in a long-axis view).
- Pulmonary artery dilation (either pulmonary artery diameter > aortic root diameter, or pulmonary artery diameter >25 mm).
- Right ventricular systolic failure: tricuspid annular plane systolic excursion (TAPSE) <18 mm .
- Systolic flattening suggests pressure overload of the right ventricle, so this is especially suggestive of pulmonary hypertension.
- Diastolic flattening suggests volume overload of the right ventricle.
- Systemic congestion (e.g., dilated inferior vena cava with reduced collapsibility).
most basic quantitative approach: systolic Pulmonary Artery Pressure (sPAP)
- PA systolic pressure = 4(tricuspid jet velocity squared) + right atrial pressure.
- sPAP <36 mm : PH is unlikely.
- sPAP 36-50 mm : Grey zone.
- sPAP >50 mm : PH is likely.
- ⚠️ Changes in sPAP over time don't necessarily track with disease improvement or deterioration. ( 36017548 )
- ⚠️ Tricuspid regurgitation is often absent in patients with proven pulmonary hypertension. (Murray 2022) Thus, the absence of a measurable tricuspid regurgitant jet doesn't exclude pulmonary hypertension by any means.

tricuspid regurgitant velocity (TRV) might be superior to the systolic pulmonary artery pressure (sPAP)
- Systolic PA pressure calculation requires estimation of the right atrial pressure, a process which is relatively inaccurate . Thus, it may be more accurate to focus solely on the tricuspid regurgitant velocity. ( 36017548 )
- Peak tricuspid regurgitant velocity <2.8 m/s : PH is unlikely
- Peak tricuspid regurgitant velocity 2.9-3.4 m/s : Grey zone.
- Peak tricuspid regurgitant velocity >3.4 m/s : PH is likely
- TRV may underestimate the pulmonary pressures due to severe tricuspid regurgitation.
- High cardiac output.
- Misinterpretation of tricuspid valve closure artifact for the TR jet.
- Incorrect assignment of the peak TR velocity due to maximal velocity boundary artifacts.
integrated approach

- The 2022 ESC guidelines recommend a combination of tricuspid valve velocity plus additional signs of pulmonary hypertension, as shown above. ( 36017548 )
- To increase the likelihood of pulmonary hypertension, signs from at least two of the following three categories should be present:
- Right ventricular dilation (RV/LV diameter ratio >1).
- Flattening of the interventricular septum.
- TAPSE/sPAP ratio <0.55 mm/mm.
- Early diastolic pulmonary regurgitation velocity >2.2 m/s.
- Right ventricular outflow tract acceleration time <105 ms and/or mid-systolic notching (the latter may indicate pre-capillary pulmonary hypertension).
- IVC diameter >21 mm with decreased inspiratory collapse.
- Right atrial dilation (end-systolic area >18 cm2).

definition of pulmonary hypertension (PH)
- PH is defined as a mPAP (mean pulmonary artery pressure) >20 mm (at rest) .
- Note that pulmonary hypertension has recently been redefined using a cutoff value of >20 mm (rather than the traditional value of >25 mm). Likewise, pulmonary arterial hypertension was redefined using a cutoff of 2 Wood units (rather than the traditional 3 Wood units).
- Patients with a mean PA pressure of 20-25 mm technically have pulmonary hypertension. However, such patients haven't been included in clinical trials on pulmonary hypertension, so there are no research-proven therapies for them. (Murray 2022)
hemodynamic evolution of PH over time
- Decompensated, advanced pulmonary hypertension is ultimately marked by a fall in cardiac output and pulmonary pressures (due to failure of the right ventricle).
- Consideration of disease severity must take into account both cardiac output and pulmonary artery pressures (not simply pulmonary artery pressures).

classification of PH
Physiological basis of ph classification.
- mPAP = PAWP + (CO)(PVR)
- This reveals mathematically that there are various reasons that the mPAP may be elevated: elevation of the PAWP, elevation of the CO, and/or elevation of the PVR.
- PH may be subcategorized based on its physiological cause:
pre-capillary PH
⬆mPAP = PAWP + (CO)( ⬆PVR )
- mPAP >20-25 mm.
- PA wedge pressure (PAWP) <15 mm. *
- PVR (pulmonary vascular resistance) >2-3 Wood units.
- PAH (pulmonary arterial hypertension).
- Group 3 PH (due to lung disease).
- Group 4 PH (CTEPH, chronic thromboembolic PH).
- Group 5 PH patients (miscellaneous).
isolated post-capillary
- ⬆mPAP = ⬆PAWP + (CO)(PVR)
- PA wedge pressure (PAWP) >15 mm. *
- PVR (pulmonary vascular resistance) <2-3 Wood units.
- Early Group 2 PH (due to left heart disease).
combined pre- & post-capillary PH
- ⬆mPAP = ⬆PAWP + (CO)( ⬆PVR )
- More advanced Group 2 PH (due to left heart disease).
- Combined left heart and lung disease overlap. ( 37026538 )
unclassified PH
⬆mPAP = PAWP + ( ⬆CO )(PVR)
- Increased airway pressures.
- Hyperthyroidism.
- Thiamine deficiency.
- Paget disease.
- AV (arteriovenous) fistula.
* PAWP cutoff of 15 mm is somewhat arbitrary. Pulmonary artery wedge pressure is an inaccurate measurement for the actual left ventricular end-diastolic pressure (figure below). Consequently, the binary division between pre-capillary and post-capillary pulmonary hypertension based on whether the pulmonary artery wedge pressure is <15 mm or >15 mm is a crude and frequently incorrect dichotomy. Additional factors should also be taken into account to distinguish pre- from post-capillary pulmonary hypertension (e.g., clinical features of PH due to left ventricular dysfunction, as discussed here: ⚡️ ). ( 36017548 ) Fluid challenge during the pulmonary artery catheterization may also help determine whether there is a component of left ventricular dysfunction.

Group 1: Pulmonary Arterial Hypertension (PAH)
- 1.1.1 Non-responders at vasoreactivity testing.
- 1.1.2 Acute responders at vasoreactivity testing.
- 1.2 Hereditary.
- Anorexigens (e.g., aminorex*, benfluorex*, dexfenfluramine*).
- Tyrosine kinase inhibitors (dasatinib*, bosutinib, ponatinib). ( 32768078 )
- Sympathomimetics : (amphetamines; cocaine; methamphetamine* especially may account for ~25% of idiopathic PH in the United States). ( 36017548 )
- Alkylating agents (e.g., cyclophosphamide, mitomycin C, busulfan, BCNU, bleomycin). These are mostly associated with pulmonary veno-occlusive disease. 📖
- Selective proteasome inhibitors (carfilzomib).
- Direct-acting antiviral agents against hepatitis C virus (sofosbuvir).
- Interferon alpha and beta.
- Solvents (trichloroethylene).
- Miscellaneous (diazoxide, leflunomide, phenylpropanolamine, Saint John's Wort).
- ~75% of patients in this category have scleroderma .
- Other causes include lupus and mixed connective tissue disease; rarely dermatomyositis and Sjogren syndrome). ( 37775164 )
- 1.4.2 HIV.
- 1.4.3 Portal hypertension.
- 1.4.4 Congenital heart disease (especially right-to-left shunt, such as atrial septal defect).
- 1.4.5 Schistosomiasis.
- 1.5 PVOD/PCH (Pulmonary veno-occlusive disease / pulmonary capillary hemangiomatosis). 📖
- 1.6 Persistent PH of the newborn.
Group 2: PH associated with left heart disease 🫀 🫀
- 2.1.1 With preserved ejection fraction. ( 36017548 )
- 2.1.2 With reduced (<40) or mildly reduced (41-49) ejection fraction.
- 2.2 Valvular heart disease.
- 2.3 Congenital/acquired cardiovascular conditions leading to post-capillary PH
Group 3: PH associated with lung disease and/or hypoxemia 🫁🫁🫁
- 3.2 Restrictive lung disease.
- 3.3 Lung disease with mixed restrictive/obstructive pattern.
- 3.4 Hypoventilation syndromes (e.g., obesity hypoventilation syndrome).
- 3.5 Hypoxia without lung disease (e.g., high altitude).
- 3.6 Developmental lung disorders (e.g., kyphosis).
Group 4: Pulmonary artery obstruction
- 4.1 Chronic thromboembolic pulmonary artery hypertension (CTEPH).
- Malignant obstruction: Angiosarcomas, other malignant tumors (e.g., renal carcinoma, uterine carcinoma, germ-cell tumors of the testis).
- Arteritis without connective tissue disease.
- Congenital pulmonary arterial stenoses.
- Hydatidosis.
- Foreign-body embolism.
Group 5: Unclear and/or multifactorial mechanisms
- Myeloproliferative disorders (e.g., chronic myelogenous leukemia, polycythemia vera, idiopathic myelofibrosis).
- Status post splenectomy.
- Sickle cell anemia.
- Sarcoidosis. 📖
- Pulmonary Langerhans cell histiocytosis.
- Neurofibromatosis type 1.
- Glycogen storage diseases.
- Gaucher disease.
- 5.4 Chronic renal failure with or without hemodialysis.
- 5.5 Pulmonary tumor thrombotic microangiopathy.
- 5.6 Fibrosing mediastinitis.
family history
- ? Connective tissue disease.
- ? Pulmonary hypertension.
- ? Clotting disorder.
review of systems
- ? Rheumatologic symptoms (Raynaud phenomenon, arthritis/arthralgia, skin changes).
- ? Symptoms of obesity hypoventilation (snoring, daytime somnolence, morning headache).
past medical history
- ? History of venous thromboembolic disease.
- ? History of heart murmur or cardiac disease.
- ? Liver disease, alcohol intake.
- ? Exposure to chemotherapy.
- ? Splenectomy.
medications/exposures – review the list above 📖
Laboratory tests.
- Complete blood count & differential cell count (? myeloproliferative disease or evidence of splenectomy).
- Liver function tests (noting, however, that congestive hepatopathy due to pulmonary hypertension can cause some abnormalities).
- If connective tissue disease suspected, also consider anticentromere antibody, anti-SCL70, anti-RNP, and anti-Ro.
- [[If scleroderma is suspected, test for anticentromere, anti-topoisomerase, anti-RNA polymerase III, and U1-RNP antibodies. (Murray 2022)]]
- HIV screen .
- TSH (thyroid stimulating hormone).
- ABG/VBG to exclude hypercapnia.
imaging studies
- Complete echocardiogram.
- Thoracic CT scan.
- Should be obtained in any patient without an evident cause of pulmonary hypertension, to exclude chronic thromboembolic pulmonary hypertension (CTEPH).
- (More on the use of VQ scan to evaluate for CTEPH here: 📖 )
other studies to consider
- PFTs (pulmonary function tests).
- Overnight oximetry or sleep study, to evaluate for obesity hypoventilation syndrome.
- Abdominal ultrasound should be performed to screen for portal hypertension.
supplemental oxygen
- Provide oxygen to achieve saturation >92%, if possible.
- Oxygen functions as a pulmonary vasodilator, so ideally hypoxemia should be avoided.
- Ambulatory oxygen may be considered when there is evidence of symptomatic benefit and correctable desaturation with exercise. ( 36017548 )
- Some patients with right-to-left shunts (Eisenmenger syndrome) won't respond to supplemental oxygen, so oxygen supplementation may be futile in such patients.
- Patients with PH tend to retain volume and become volume overloaded. Avoiding fluid retention is one of the key objectives in managing patients with PH. ( 36017548 )
- Diuretic resistance may result from poor renal perfusion, renal venous congestion, and intense neurohormonal activation. Combination therapy may be needed, including loop diuretics, thiazides, and mineralocorticoid receptor antagonists.
medications to avoid
- Nasal decongestants.
- Nitrates, ACE inhibitors, ARBs (angiotensin receptor blockers).
- Beta-blockers.
lung transplantation
- Lung transplantation may be a consideration for pulmonary arterial hypertension that fails to respond to therapy.
- Indications to refer for transplantation evaluation are listed here: 📖
(anticoagulation)
- Some registry data has suggested improved outcomes among patients with idiopathic pulmonary arterial hypertension (IPAH) who were anticoagulated. However, data is contradictory and of low quality (with anticoagulation carrying obvious risks).
- Currently guidelines make no recommendations regarding anticoagulation for pulmonary hypertension. ( 36017548 )
when to perform vasoreactivity testing
- 1.1 (idiopathic pulmonary arterial hypertension).
- 1.2 (hereditary pulmonary arterial hypertension).
- 1.3 (drug-induced pulmonary arterial hypertension).
- Only patients in these groups may have a durable clinical response to calcium channel blockers. Consequently, these patients are the only ones where vasoreactivity testing would change clinical management. (Murray 2022)
contraindications to vasoreactivity testing
- Overt right ventricular failure (such patients aren't candidates for calcium channel blocker therapy).
- Vasoreactivity testing for identifying candidates for calcium channel blocker therapy is not recommended in patients known to have pulmonary arterial hypertension of other types (1.4-1.6), or PH groups 2-5. (Class III recommendation, 36017548 )
how to perform vasoreactivity testing
- May use short-acting IV epoprostenol sodium or inhaled nitric oxide.
- Nitric oxide is the preferred pulmonary vasodilator (e.g., 20 ppm for 5-10 minutes). Inhaled nitric oxide is easier to use than IV epoprostenol, because nitric oxide titration may be performed more rapidly.
three criteria required to define a positive vasodilator response:
- mPAP falls by >10 mm.
- mPAP reaches a value <40 mm.
- Cardiac output is stable or increases. ( 36017548 )
implications of positive vasoreactivity
- This is a favorable prognostic sign.
- Patients can be treated with calcium channel blockers.
indications for oral calcium channel blockers (CCBs)
- In other forms of PAH, even a favorable vasodilator challenge doesn't predict a durable, long-term response to calcium channel blockers. ( 36017548 )
- At baseline, only 12% of patients have vasoreactivity. ( 37026538 )
- Among patients with vasodilator responsiveness, only about half will obtain long-term benefit from calcium channel blockers (the other half of patients will lose responsiveness over time).
- ⚠️ If calcium channel blocker therapy doesn't improve the patient to NYHA functional class I or II, then additional therapy should be utilized.
- ⚠️ Long-term response to CCB therapy is uncommon, so patients need to be monitored for disease progression. ( 23809320 )

adverse effects of calcium channel blockers
- Hypotension is common.
- Peripheral edema.
- Hypoxemia due to impaired ventilation-perfusion matching.
- Right ventricular failure (if diltiazem is utilized).
preferred agents & dose
- Agents: Amlodipine, felodipine, or nifedipine may be the best agents (since these provide vasodilation, without negative inotropic effects).
- Amlodipine : Start 5 mg daily, target dose 15-30 mg daily.
- Nifedipine : Start at 10 mg TID (or nifedipine XR 30 mg/daily), target dose 20-60 mg BID-TID (or nifedipine XR 120-240 mg daily).
- Felodipine : Start 5 mg daily, target dose 15-30 mg daily.
Endothelin-1 is a vasoconstrictor and smooth muscle mitogen that increases pulmonary vascular resistance. Similar effects seem to result from either selective inhibition of endothelin A, or nonselective inhibition of both endothelin A and B.
bosentan (nonselective endothelin A & B antagonist)
- Studied in patients with idiopathic PAH and connective tissue-associated PAH who were functional class III-IV.
- Improves exercise capacity, functional class, hemodynamics, and time to clinical worsening. ( 36017548 )
- Black box warning due to hepatotoxicity. Liver function tests should be followed closely, with discontinuation if AST/ALT >3 times upper limit normal with symptoms or elevated bilirubin. Roughly 10% of patients need to discontinue bosentan due to abnormal liver tests. ( 36017548 )
- Avoid abrupt withdrawal.
- Can cause anemia (it usually decreases hemoglobin by about 1 g/dL). (Murray 2022)
- Metabolized by the CYP system (inducer and substrate of CYP 2C9 and 3A4). Interactions include reducing serum levels of sildenafil and tadalafil – which impairs the ability to combine bosentan with phosphodiesterase-5 inhibitors. ( 36017548 )
- Teratogenic, cannot be used in pregnancy (class effect).
- Starting dose 62.5 mg BID.
- Target dose: 125 mg BID.
ambresartan (selective inhibitor of endothelin A)
- Approved for pulmonary arterial hypertension (PAH) class II-III.
- Demonstrated to improve symptoms, exercise capacity, hemodynamics, and time to clinical worsening. ( 36017548 )
- Unlike bosentan, there is no increase in abnormal liver function tests. ( 36017548 )
- Doesn't appear to be hepatotoxic.
- Can cause anemia.
- Initial dose 5 mg daily.
- Target dose 10 mg daily.
macitentan (dual endothelin receptor antagonist)
- Shown to improve exercise capacity and reduce clinical worsening in pulmonary arterial hypertension. ( 36017548 )
- Doesn't appear to be hepatotoxic (it was beneficial for patients with portopulmonary hypertension in the PORTICO trial). (PORTICO trial 31178422 )
- Anemia may occur in ~4% of patients. ( 36017548 )
- Initial dose 10 mg daily, this is also the target dose.
- Both phosphodiesterase-5 inhibitors (sildenafil, tadalafil) and guanylate cyclase stimulators (riociguat) act on the same molecular pathway, to cause vasodilation of the pulmonary vasculature. Phosphodiesterase-5 inhibitors enhance the activity of endogenous nitric oxide, whereas guanylate cyclase stimulators may directly cause pulmonary vasodilation in the absence of nitric oxide (allowing guanylate cyclase stimulators to be independently effective, regardless of prevailing nitric oxide levels).
- Phosphodiesterase-5 inhibitors and guanylate cyclase stimulators (riociguat) cannot be combined (since this may cause systemic hypotension). ( 36017548 )
indications & side effects
- Shown to improve exercise capacity, symptoms, and hemodynamics in patients with pulmonary arterial hypertension. ( 36017548 )
- Side effects are generally mild/moderate (e.g., headache, flushing, and epistaxis).
- Seems to be similar to sildenafil, but longer half-life may provide more sustained action.
- One RCT of patients with pulmonary arterial hypertension demonstrated improvements in exercise capacity, hemodynamics, symptoms, and time to clinical worsening. ( 19470885 )
- An RCT of patients with pulmonary arterial hypertension demonstrated improvements in exercise capacity, hemodynamics, functional classification, and time to clinical worsening. ( 23883378 )
- The REPLACE trial demonstrated that patients on phosphodiesterase-5 inhibitor therapy (mostly sildenafil) improved when switched to riociguat at 2.5 mg TID. ( 33773120 )
- Side-effect profile is similar to that of phosphodiesterase-5 inhibitors.
contraindications/risks
- Impaired ventilation/perfusion matching may impair oxygenation.
- Avoid co-administration with nitrates (may cause severe hypotension).
- Riociguat is teratogenic.
dose & pharmacology
- Initial dose and target dose are 20 mg TID. (However, higher doses were found to be more effective in the SUPER-1 trial. Such doses may occasionally be utilized in practice.) ( 16291984 )
- Half-life is ~3-4 hours.
- Starting dose is 20-40 mg daily, target dose is 40 mg daily.
- Half-life in pulmonary hypertension is ~35 hours.
- Starting dose is 1 mg TID, target dose is 2.5 mg TID.
- Half-life is 7 hours, but may be prolonged to 12 hours in patients with pulmonary artery hypertension.
basics – mechanism
- Epoprostenol and treprostinil are prostacyclin analogues , whereas selexipag is an orally available prostacyclin receptor agonist (which is chemically distinct from prostacyclin).
- Pulmonary vasodilation.
- May reduce pulmonary vascular remodeling due to antiplatelet aggregation effects.
indications
- IV epoprostenol is generally considered to be the most powerful agent for PAH. It has been shown to improve hemodynamics, functional capacity, and survival (among patients with pulmonary arterial hypertension). ( 8532025 )
- IV epoprostenol is often the preferred option for the highest risk patients.
- Improves hemodynamic parameters, symptoms and exercise capacity in Group I PAH.
dosing of IV epoprostenol for chronic PH
- May titrate hourly as tolerated to a target dose of ~12-20 ng/kg/min.
- The typical dose range after one year of therapy is 16-30 ng/kg/min, with wide individual variability. ( 36017548 )
- Half-life is 3-5 minutes, so systemic levels will change almost immediately in response to dose adjustments. Metabolism occurs rapidly in the blood.
- Induced metabolism requires a continuous increase in dose to maintain symptom control. (Fishman 2023)
dosing of IV epoprostenol for acute right heart failure in the ICU
- Acute-on-chronic deterioration in a patient with known Type 1 PH.
- Amniotic fluid embolism with refractory RV failure (if ECMO is unavailable). ( 33417901 )
- ⚠️ This should generally be regarded as a last-line therapy , which ideally should be avoided. Specifically, either one or a combination of inhaled epoprostenol plus inhaled nitric oxide is generally safer and preferable.
- Systemic hypotension.
- Nausea and vomiting.
- Thrombocytopenia and impaired platelet function.
- Hypotension may occur due to the effect of epoprostenol on systemic vascular resistance. If there are no other treatment options, this may be alleviated with the use of a vasopressin infusion to increase the systemic vascular resistance.
- Hypoxemia may be dose-limiting if it is treatment refractory. (Note that patients would typically already be on an aggressive regimen of inhaled pulmonary vasodilators prior to initiation of intravenous epoprostenol.)
- Start at 2 ng/kg/min. The dose may be up-titrated by 2 ng/kg/min every 15 minutes until the patient stabilizes, or side-effects occur. ( 19332472 )
- IV epoprostenol is preferable to treprostinil in this situation, because the short half-life of epoprostenol allows for more prompt titration. If epoprostenol causes destabilization, it may be discontinued with subsequent drug clearance within minutes.
dosing of treprostinil (SC or IV)
- Start at 1.25 ng/kg/min.
- Dose is determined by tolerability and effectiveness.
- Typical dose range at one year is 25-60 ng/kg/min, with wide individual variability. ( 36017548 )
- Half-life is ~4 hours, so titration will take much longer to reach steady state (as compared to epoprostenol). Metabolism occurs in the liver, predominantly by CYP2C8.
side effects & risks of epoprostenol/treprostinil
- Hypoxemia may result from diffuse pulmonary vasodilation, which impairs ventilation-perfusion (V/Q) matching. ( 32654737 )
- Hypotension .
- Thrombocytopenia and reduced platelet function .
- Catheter-related sepsis.
- Deep vein thrombosis.
- Pump dysfunction or kinking may lead to abrupt discontinuation (which may cause hemodynamic decompensation).
- Headache, jaw pain, musculoskeletal aches and pains (mostly in the legs and feet).
- Diarrhea, nausea, vomiting.
- Blotchy erythematous rash.
- Acute overdose may cause systemic hypotension.
- Chronic overdose may cause development of a hyperdynamic state with high-output heart failure.
- Selexipag is an oral prostacyclin receptor agonist.
- When given alone or combined with an endothelin receptor antagonist and/or phosphodiesterase-5 inhibitor, selexipag reduced the risk of composite morbidity or mortality events in one Phase III RCT by 40%. (GRIPHON trial, 26699168 ) Most subjects in this trial were already on background medication (including 15% with an endothelin-receptor agonist, 32% with a phosphodiesterase-5 inhibitor, and 33% on both an endothelin-receptor agonist and a phosphodiesterase-5 inhibitor).
- Unfortunately, the addition of selexipag failed to cause any improvement when given on top of dual therapy with macitentan plus tadalafil. (TRITON trial, 34593120 )
side effects & risks
- The most common side effects are headache, diarrhea, nausea, and jaw pain (similar to those of prostacyclin analogs).
- Dose is initially 200 ug BID, with gradual up-titration as high as can be tolerated (up to 1600 ug BID).
The initial approach to patients depends on risk stratification. Patients can be divided into three groups:

Very occasional patients in groups 1.1-1.3 (idiopathic, inheritable, or drug-associated pulmonary arterial hypertension) have a positive response to vasodilation therapy during PA catheterization. Such patients may be treated with oral calcium channel blockers as described above: 📖
Most patients will not be candidates for calcium channel blocker therapy. Such patients are managed using the algorithm shown below.

additional considerations based on the specifics of PAH
Pah with cardiopulmonary comorbidities.
- Left heart phenotype : Elderly patients with clinical features suggestive of Group 2 PH (such features are discussed further here: ⚡️ ).
- Cardiopulmonary phenotype : Elderly, mostly male patients with hypoxemia, smoking history, and risk factors for left heart disease.
- Older patients with comorbidities are less likely to benefit from pulmonary vasodilator medications. Such patients are often excluded from trials of pulmonary vasodilators, rendering management challenging.
- Monotherapy with an oral phosphodiesterase-5 inhibitor or endothelin receptor antagonist should be considered. However, among patients with comorbidities, these agents may have increased risks of precipitating left heart failure or exacerbating hypoxemia (due to impaired ventilation/perfusion matching).
PAH associated with drugs and toxins
- In patients with a low-risk profile, the initial management may be to discontinue the causative agent and follow for 3-4 months. In patients with intermediate or high-risk profile, treatment for pulmonary arterial hypertension should start immediately.
- Some patients may improve over time, allowing for de-escalation of therapy.
PAH associated with connective tissue disease
- Pulmonary arterial hypertension.
- Left heart dysfunction.
- Interstitial lung disease.
- Pulmonary emboli.
- Thus, every patient must be evaluated to determine the components of their pulmonary hypertension.
- If the interstitial lung disease is severe and felt to the cause of pulmonary hypertension, then treatment may resemble that of Group 3 PH. 📖
- If the interstitial lung disease is mild in comparison to the pulmonary hypertension and no other etiology of pulmonary hypertension is discovered, this implies predominantly Group 1 PH. Such patients may benefit from PAH-specific treatment (as discussed in the section above).
- HIV may cause a form of Group I pulmonary artery hypertension (PAH) that is pathologically indistinguishable from idiopathic PAH.
- Clinical features are similar to non-HIV idiopathic PAH.
- Among patients with PAH and HIV, PAH is a common cause of death (this isn't merely an incidental finding).
epidemiology
- The annual incidence PAH in HIV might be ~0.25-0.5%. Echocardiographic data suggest that the prevalence could be on the order of 5-10%. (Murray 2022)
- There is no correlation between the severity of PAH and the stage of HIV, or the level of immunodeficiency. ( 36017548 )
differential diagnosis includes
- Sympathomimetic use (especially methamphetamine).
- Antiretroviral therapy will reduce HIV replication and immune dysregulation, which may delay progression. Additionally, antiretroviral therapy may actually have a beneficial effect on hemodynamics.
- Treatment is generally similar to that of idiopathic PAH. However, there may be increased risks of drug interactions (e.g., with antiretroviral medications). Consequently, initial monotherapy is generally recommended, with individualized escalation to combination therapy if patients don't respond adequately. ( 36017548 )
- Portopulmonary hypertension seems to occur due to impaired hepatic metabolism of vasoconstrictive agents (e.g., thromboxanes, serotonin, bradykinin, and neuropeptide Y).
- Pulmonary arterial lesions in portopulmonary hypertension are histologically indistinguishable from those seen in idiopathic pulmonary arterial hypertension. ( 30526986 )
epidemiology: causes of pulmonary hypertension
- PH occurs in ~2% of patients with cirrhosis.
- PH is detected in ~5% of patients undergoing liver transplantation evaluation. (Murray 2022)
- PH is usually diagnosed ~4-7 years after the diagnosis of portal hypertension.
- Portopulmonary hypertension doesn't correlate with the severity of the liver disease. (Murray 2022)
- [2] Portosystemic shunt of other etiologies: Portopulmonary hypertension may rarely develop in the absence of any liver disease (e.g., congenital extrahepatic cavoportal shunts). ( 36017548 )
- Initially, symptoms are often related to portal hypertension.
- Exertional dyspnea.
- Syncope, chest pain.
- Features of right heart failure may be difficult to distinguish from features of cirrhosis (e.g., edema, abdominal distension). (Murray 2022)
definition of portopulmonary hypertension & evaluation
- Portopulmonary hypertension is defined as pre-capillary pulmonary hypertension in patients with portal hypertension or a portosystemic shunt, in the absence of alternative explanations (i.e., sPAP >20 mm, PVR >2 Wood units, and PA wedge pressure <15 mm).
- (1) Elevated cardiac output may increase the tricuspid regurgitant jet velocity, which may lead to overestimation of pulmonary artery pressures on echocardiography. PA catheterization may demonstrate that the sPAP is not actually elevated – so there isn't any pulmonary hypertension at all.
- (2) Many patients with cirrhosis do have truly elevated sPAP >20 mm. However, this elevation is due to increased cardiac output , rather than an elevation of their PVR (pulmonary vascular resistance). Such patients do not have portopulmonary hypertension, but instead they are regarded as having “unclassified PH.” Management involves follow-up, but not treatment with medications for pulmonary arterial hypertension.
management of portopulmonary hypertension
- In patients with portopulmonary hypertension and mild liver disease, pulmonary hypertension may be a major cause of death.
- Initial monotherapy should be considered more often, followed by combination therapy if necessary. ( 31178422 )
- An RCT dedicated to portopulmonary hypertension involved macitentan, which demonstrated improvement in pulmonary vascular resistance but no differences in functional class, 6-minute walk distance, or NT-BNP levels. (PORTICO trial 31178422 ) A single-arm observational study likewise found benefit from ambrisartan. ( 32008947 )
- Uncontrolled series have consistently found that prostacyclin infusions improve hemodynamics among patients with portopulmonary hypertension. This is probably the most powerful approach to optimize patients and bridge them to liver transplantation (discussed further below). (Fishman 2023)
- Diuretics are especially important, because both portopulmonary hypertension and cirrhosis may promote sodium retention. (Fishman 2023)
- ⚠️ Calcium channel blockers should be avoided, because they can worsen splanchnic vasodilation. ( 27326810 )
- ⚠️ Beta-blockers are often used in cirrhosis, but beta-blockers should be avoided as these can worsen cardiac output. ( 27326810 )
- ⚠️ TIPS placement is contraindicated in severe pulmonary hypertension. TIPS may reduce liver perfusion, causing reduced metabolism of endogenous pulmonary vasoconstrictors (e.g., endothelin).
portopulmonary hypertension & liver transplantation
- The response of pulmonary hypertension to liver transplantation is variable. Pulmonary hypertension may gradually improve following transplantation, but severe pulmonary hypertension can be progressive.
- mPAP >50 mm correlated with 100% perioperative mortality.
- mPAP 35-50 mm with PVR >3 Wood unit correlated with 50% perioperative mortality.
- (#1) mPAP <35 mm with PVR <5 Wood units.
- (#2) mPAP 35-45 mm with PVR <3 Wood units.
- ⚠️ mPAP >45 mm is regarded as an absolute contraindication to liver transplantation.
- If pulmonary hypertension precludes a liver transplantation, medical therapy should be utilized to control pulmonary hypertension. If hemodynamics respond well to therapy, then transplantation may be reconsidered.
clinical classification of pulmonary arterial hypertension associated with congenital heart disease
- Eisenmenger syndrome includes all patients with systemic-to-pulmonary shunts that progress to severely elevated PVR and undergo subsequent reversal of the shunt (or bidirectional shunting). Eisenmenger syndrome is an ominous sign overall, but it implies that the right ventricle has preserved systolic function (as required to generate the pressure required to shunt blood towards the left side of the heart).
- Cyanosis, hypoxemia.
- Secondary erythrocytosis.
- Hemoptysis.
- Coagulation abnormalities, including thrombocytopenia.
- Brain abscess, ischemic stroke.
- PVR is mild-moderately increased, and systemic-to-pulmonary shunting is still prevalent.
- Eisenmenger syndrome has not occurred (there is no reversal of the shunt), so there is not cyanosis at rest.
- Some patients may be eligible for shunt correction.
- Markedly elevated PVR in the presence of cardiac defects considered to be nonsignificant (usually ventricular septal defect <1 cm or atrial septal defects <2 cm).
- Clinical picture and therapy may be similar to that of idiopathic pulmonary arterial hypertension.
- Closure of shunts is contraindicated.
- Congenital heart disease is repaired, but PAH either persists or recurs.
diagnosis and evaluation
- PA catheterization with compartmental oximetry is required to calculate the ratio of blood flow through the pulmonary circulation in comparison to the systemic circulation (Qp/Qs).
- Thermodilution should be avoided in the presence of intracardiac shunts.
- Shunt closure may be considered in patients with lower pulmonary pressures (e.g., PVR <3-5 Wood units). However, once significant pulmonary arterial hypertension has developed, it's generally too late to close the defect. Precise indications for closure are beyond the scope of this chapter.
- There should be a low threshold to consider infection, since these patients are at increased risk of endocarditis and brain abscess.
- Supplemental oxygen should be utilized in patients where it causes significant improvement. However, patients with shunt physiology may not respond to supplemental oxygen.
- Endothelin receptor antagonist bosentan has been shown to improve exercise capacity in patients with Eisenmenger syndrome. ( 16801459 ) It's conceivable that endothelin receptor antagonists could constitute a preferred oral therapy for these patients.
- In patients with pulmonary-to-systemic shunting, placement of an indwelling line for epoprostenol infusion may create a risk of cerebral embolic events. Consequently, there may be some advantages to subcutaneous treprostinil.
- (Closing the shunt is contraindicated, since this would precipitate volume overload of the right ventricle).
- For patients with prevalent systemic-to-pulmonary shunting ( prior to the development of Eisenmenger syndrome), the benefit of PAH therapies is less well established. ( 36017548 ) Reduction of the pulmonary vascular resistance could increase the shunt fraction.
- Pulmonary capillary hemangiomatosis may represent a secondary phenomenon that results from capillary congestion, as a result of pulmonary veno-occlusive disease.
- Obstruction of post-capillary venules with fibrous tissue that may cause complete occlusion (PVOD).
- Capillary dilation and proliferation (PCH).
- 💡 CT scan reveals features of cardiogenic pulmonary edema , but there is no left ventricular dysfunction.
- 💡 Patient with PAH develops cardiogenic pulmonary edema after initiation of pulmonary vasodilators .
general epidemiology
- PVOD/PCH is ~1/10th as common as idiopathic PAH, so it is quite rare.
- PVOD/PCH may occur in siblings (due to recessive genetics involving the EIF2AK4 gene).
- Inheritable cases tend to present earlier (~25 years old), whereas acquired cases often present later (~60 years old). ( 34295399 )
most cases are idiopathic , but PVOD/PCH may be associated with:
- PVOD/PCH may tend to present ~3-4 months following transplantation. (Murray 2022)
- The incidence seems to be rare, largely limited to case reports. (Fishman 2023) However, the diagnosis may be under-recognized, since evidence of PVOD/PCH is found in ~1/3 of autopsies performed >1 year after transplantation. (Murray 2022)
- Alkylating agents, including cyclophosphamide, mitomycin C, bleomycin, busulfan, gemcitabine, and carmustine (BCNU).
- Scleroderma 📖 (and perhaps also lupus).
- Organic solvents (e.g., trichloroethylene).
clinical presentation
- Dyspnea is the most common symptom.
- Hemoptysis may occur in ~30% of patients.
- Hemorrhagic pleural effusion . ( 28991555 )
- Orthopnea may occur (which can mimic left-sided heart failure).
- Digital clubbing may be seen. ( 36017548 )
- If PVOD/PCH isn't recognized initially, ~50% of patients may develop cardiogenic pulmonary edema after initiation of a pulmonary vasodilator.
radiologic features suggestive of PVOD/PCH:
- Interlobular septal thickening.
- Pleural effusions.
- Ground-glass opacification (may be diffuse, mosaic, or patchy).
- (b) Normal left atrial size.
- (2) Centrilobular ground-glass micronodules (although these may also be seen in idiopathic pulmonary arterial hypertension).
- (3) Mediastinal lymphadenopathy may be caused by vascular congestion. ( 36017548 )
differential diagnosis of PH plus centrilobular nodules:
- PVOD/PCH (as discussed above).
- Pulmonary tumor emboli . 📖
- Nodules may be better defined than in PVOD/PCH.
- There is an absence of associated interlobular septal thickening or pleural effusion.

- VQ scan may be normal.
- VQ scan may reveal multiple small areas of hypoperfusion. These shouldn't be misinterpreted as evidence of CTEPH (chronic thromboembolic pulmonary hypertension).
PA catheterization
- PVOD/PCH causes precapillary pulmonary hypertension (mPAP >20 mm, PAWP <15 mm, PVR <2 Wood units).
- This may tend to mimic idiopathic pulmonary arterial hypertension (iPAH).
- Vasodilator testing may induce cardiogenic pulmonary edema. If PVOD/PCH is suspected, this is contraindicated. ( 34295399 )
confirmation of the diagnosis
- Genetic studies revealing biallelic EIF2AK4 mutations confirms the diagnosis of inheritable PVOD/PCH.
- Lung biopsy is high-risk and not generally recommended. ( 36017548 )
- General supportive measures for pulmonary hypertension may be utilized.
- Treatments for PAH are often ineffective and may cause deterioration (by precipitating pulmonary edema). Nonetheless, some experts may favor a very cautious trial of vasodilator therapy as a bridge to lung transplantation. ( 34022029 )
- Lung transplantation is the only definitive treatment. Any patients who are potentially transplant candidates should be referred urgently. ( 34022029 ) The disease course of PVOD/PCH is generally more aggressive than idiopathic pulmonary arterial hypertension, with a one-year mortality of ~70%. ( 34743853 )
- Group 2 PH is the most common form of PH (accounting for ~75% of cases).
clinical features that suggest PH is due to left heart disease
- Age >60-70 years old.
- Hypertension.
- Dyslipidemia.
- Glucose intolerance / diabetes mellitus.
- Atrial fibrillation.
- Coronary artery disease.
- Prior cardiac intervention.
- LAD (left axis deviation).
- LVH (left ventricular hypertrophy).
- LBBB (left bundle branch block).
- Left atrial maximal area >27-30 cm2. 📖 ( 29530618 )
- Left atrial dilation (LAVI >34 ml/m2).
- Left ventricular hypertrophy.
- Significant (> grade 1) diastolic dysfunction. ( 36017548 , 37775164 )
hemodynamic forms of group 2 PH
- mPAP >20 mm.
- PA wedge pressure (PAWP) >15 mm.
- This occurs initially in the course of left heart failure. It is the most common form of PH due to left heart disease.
- PH is simply due to passive congestion .
- This represents disease progression from isolated post-capillary PH (due to pulmonary vascular remodeling).
- (Further discussion of hemodynamic profiles in PH above: 📖 )
evaluation of pulmonary hypertension
- In the presence of predominant left heart disease and mild pulmonary hypertension, further evaluation may be unnecessary.
- Otherwise, a full evaluation should be undertaken – especially if right ventricular dysfunction seems disproportionately severe in comparison to the degree of left heart disease.
- Evaluation may help exclude additional causes of PH (e.g., chronic thromboembolic pulmonary hypertension). ( 36017548 )
- Medications that appear to reduce pulmonary pressures include: hydralazine plus isosorbide dinitrate, SGLT2 inhibitors (sodium-glucose cotransporter 2 inhibitors), and sacubitril/valsartan. ( 21632515 , 36813291 )
- Prostacyclins are contraindicated (increased mortality in the FIRST trial). ( 9266782 )
- Endothelin antagonists contraindicated. For example, bosentan increased heart failure exacerbations in the ENABLE trial. Even among patients with combined pre- and post-capillary pulmonary hypertension, macitentan worsened fluid retention and functional class in the MELODY-1 trial. ( 29437943 )

- 3.6 Developmental lung disorders (e.g., kyphosis).
- Severe PH here is defined as PVR (pulmonary vascular resistance) >5 Wood units. ( 36017548 )
- Severe PH occurs in ~1-5% of patients with COPD and <10% of patients with advanced ILD. ( 36017548 )
- Exacerbations of lung disease may transiently increase pulmonary pressure, so evaluation should ideally be performed when patients are clinically stable.
- Initial evaluation includes echocardiography and contrast-enhanced CT scan.
- Assessment for lung transplantation.
- Suspicion of pulmonary artery hypertension (PAH) as the etiology of pulmonary hypertension (rather than underlying lung disease).
differential diagnosis
- Chronic pulmonary disease (e.g., COPD, IPF) often occurs in elderly patients with significant comorbidity. Age and comorbidity increase the rate of pulmonary hypertension due to left heart failure (Group 2 pulmonary hypertension).
- Connective tissue disorders cause a variety of etiologies of pulmonary hypertension, as discussed further above. 📖
- 💡 Even in patients with a seemingly obvious cause of PH, complete evaluation should be performed to evaluate for alternative etiologies (as explored above).
management
The cornerstone is treating the underlying lung disease .
- Hypoxemia management: Supplemental oxygen sufficient to prevent hypoxemia may substantially improve pulmonary pressures.
- Hypercapnia management : Noninvasive ventilation may be beneficial for some patients (e.g., obesity hypoventilation syndrome).
- Disease-specific therapies should be optimized.
- Lung transplantation may be considered for selected patients.
inhaled prostacyclin analogues
- Since inhaled prostacyclins are distributed via inhalation to the best ventilated alveoli, they may improve ventilation-perfusion matching (V/Q matching), thereby improving oxygenation and ventilation. Inhaled prostacyclins may also simultaneously improve pulmonary pressures.
- Inhaled prostacyclins may be especially useful for patients with lung disease (e.g., Group III pulmonary hypertension), in whom systemic prostacyclins cause impaired ventilation-perfusion matching and thus worsened hypoxemia.
- Inhaled prostacyclins may also theoretically avoid systemic side effects.
- The INCREASE trial demonstrated that among a population of patients with PH due to interstitial lung disease, inhaled treprostinil at a dose of 72 ug QID led to improved 6-minute walk distance and reduced clinical worsening. ( 33440084 , 34767495 )
- The TRIUMPH-1 trial demonstrated that inhaled treprostinil improved 6-minute walk distance and quality of life among patients on a background of therapy with either an endothelin-receptor antagonist or phosphodiesterase 5-inhibitor. ( 20430262 )
- However, current treatment algorithms don't generally recommend the addition of inhaled prostacyclin analogues in this fashion.
other advanced therapies for PAH
- These should not be utilized, since generalized pulmonary vasodilation will impair ventilation/perfusion matching and thereby may exacerbate hypoxemia. This may cause deterioration among patients with substantial underlying hypoxemia and/or hypercapnia.
- For example, bosentan was demonstrated to worsen hypoxemia and quality of life among patients with COPD. ( 18448495 ) Riociguat appeared to increase mortality among patients with idiopathic interstitial pneumonia. (RISE-IIP study, 31416769 )
- Chronic thromboembolic pulmonary disease (CTEPD) : Patients with symptoms attributed to post-thromboembolic obstructions within the pulmonary arteries.
- Chronic thromboembolic pulmonary hypertension (CTEPH) : A subset of patient with CTEPD who have pulmonary hypertension.
- CTEPH is the only form of PH that can potentially be cured without lung transplantation. Therefore, it is important to always consider the possibility of CTEPH whenever evaluating for the etiology of pulmonary hypertension.
- Elevated pulmonary pressures lead to further vascular remodeling and worsening of pulmonary pressures – thus forming a vicious spiral that leads to ongoing deterioration.
- CTEPH might occur in ~2% of patients with PE, but estimates vary widely.
- Permanent intravascular devices (pacemaker, long-term central lines, especially veno-arterial shunts).
- Recurrent venous thromboembolic disease.
- Splenectomy.
- Antiphospholipid syndrome.
- Inflammatory bowel disease.
- Essential thrombocythemia, polycythemia vera.
- Thyroid hormone replacement.
- Malignancy.
- About half of CTEPH patients have no known history of pulmonary embolism.
- The most common symptoms are fatigue and dyspnea on exertion.
echocardiogram
- Pulmonary embolism with estimated systolic PA pressure >60 mm should raise suspicion of CTEPH (acute PE shouldn't cause this degree of pulmonary hypertension without precipitating cardiovascular collapse; this degree of pulmonary hypertension implies chronicity with right ventricular hypertrophy ). ( 36017548 )
CT scan findings
Pulmonary arteries.
- Most patients have multiple, bilateral arterial abnormalities. (Walker 2019)
- [1] Clot is more plaque-like , with a tendency to hug the arterial wall. Clot may have a crescent-shaped configuration that forms obtuse angles with the pulmonary arterial wall. (Shepard 2019)
- (#1) Initially, a central “dot” of contrast may be surrounded by circumferential thrombus.
- (#2) Ongoing recanalization may cause the walls of the artery to appear irregularly thickened .
- [3] Intravascular web or flap (linear filling defect).
- [4] Stenosis : It is a sign of CTEPH if the vessel is contracted , causing it to be smaller than corresponding arteries in the contralateral lung. (Shepard 2019) Complete vessel occlusion may occur. Post-stenotic dilation may occur.
- [5 ] Calcification of the thrombus may occur.
mosaic perfusion
- Areas of hypoattenuated lung are associated with reduced pulmonary artery size.
- Enlarged bronchial arteries (>1.5 mm in diameter).
other findings
- Peripheral lung opacities due to prior pulmonary infarction.
- Bronchial dilation may occur in areas with severely stenotic or occluded pulmonary arteries. (Shepard 2019)
- Bronchial arteries may be unusually prominent.
performance of CT scan for CTEPH
- Performance of CTA for CTEPH is limited. It is reported as sensitivity of 76% and specificity of 96%. ( 36017548 ) Distal disease may be missed by CT scan.
- However, performance may be superior with modern, multi-detector CT scanners.
- VQ scan is the test of choice for the diagnosis of CTEPH (sensitivity ~90-100%, specificity of 94-100%). VQ scan has a higher performance than CT angiography.
- However, VQ abnormalities in CTEPH may be less dramatic than those seen in acute PE (due to partial recanalization of pulmonary arteries). (Murray 2022)
- IPAH (idiopathic pulmonary arterial hypertension) or PVOD/PCH (pulmonary veno-occlusive disease/pulmonary capillary hemangiomatosis) may cause multiple small, subsegmental perfusion defects.
- Extrinsic vascular compression due to lymphadenopathy, fibrosing mediastinitis, or tumor.
- Pulmonary vasculitis.
- Pulmonary artery sarcoma.
- Prior PE with residual thrombus, in the absence of pulmonary hypertension.
- A retrospective case series found an increased rate of recurrent thromboembolism rates among patients treated with DOACs. ( 31557382 , 35305871 )
- Evaluation for antiphospholipid syndrome should be performed at the time of CTEPH diagnosis. ( 36017548 ) Patients with antiphospholipid syndrome require anticoagulation with warfarin, rather than direct oral anticoagulants. ( 36813291 )
- Surgical thromboendarterectomy may be curative. This is the treatment of choice, if possible. Consultation should be obtained with a CTEPH surgical center of excellence (e.g., University of California at San Diego ). CTEPH surgery should ideally be performed at a high-volume center (>50 surgical procedures per year). ( 36017548 )
- Treatments for pulmonary arterial hypertension may be beneficial for patients who aren't candidates for surgery, or patients with persistent pulmonary hypertension despite surgery.
- Available studies support the use of riociguat, s.c. treprostinil, or macitentan. Riociguat may be the single preferred oral agent at this point in time. ( 36017548 ) Although off-label, severe PH can be treated with combination therapies (e.g., combination therapy involving a phosphodiesterase-5 inhibitor or riociguat, plus an endothelin receptor antagonist). The pathophysiology of arteriole changes in CTEPH is similar to idiopathic PAH, so a similar treatment regimen may make sense.
- Balloon pulmonary angioplasty is an interventional option, although long-term outcomes remain unclear.
- Lung transplant may be considered if emboli are too distal to remove surgically.
- PH may occur in ~10% of patients with stable sickle cell disease (using a cutoff of mPAP >25 mm). These patients seem to be roughly divided between pre-capillary and post-capillary pulmonary hypertension.
- Increased age.
- Renal insufficiency.
- Severity of hemolytic anemia (e.g., lower hemoglobin, higher lactate dehydrogenase; higher bilirubin). Hemolysis seems to be mechanistically related to pulmonary hypertension. (Fishman 2023)
screening echocardiography & NT-proBNP
- Adults with Sickle cell disease should be screened with echocardiography and measurement of NT-proBNP values.
- Screening should be done when patients are at steady state (not during a vaso-occlusive crisis).
right heart catheterization
- A general approach to PH on echocardiography is shown above (with further discussion here: 📖 ).
- (1) Tricuspid regurgitation velocity >3 m/s.
- (2) Tricuspid regurgitation velocity >2.5 m/s plus NT-proBNP >16 pg/ml AND 6-minute walk distance <333 meters.
- Pulmonary artery catheterization may confirm or refute the diagnosis of pulmonary hypertension, as well as determine the type of pulmonary hypertension. 📖
evaluation for other etiologies of PH
- Patients found to have PH should undergo a complete evaluation for alternative causes of PH (discussed above: 📖 ).
- Pulmonary thromboemboli may be a significant contributor. Patients should be evaluated for chronic thromboembolic pulmonary hypertension (CTEPH).
- Diastolic heart failure is common.
basic supportive measures:
- Hydroxyurea.
- Exchange transfusions.
- Chronic hypoxemia should be treated with oxygen supplementation.
- Nocturnal hypoxemia.
- Thromboembolic disease.
- Left ventricular disease.
PAH medications for patients with precapillary PH:
- There is a lack of evidence to support these in sickle cell disease.
- Sildenafil was found to be ineffective among patients with sickle cell disease and elevated tricuspid regurgitant velocity. Sildenafil appeared to increase hospitalization rates for painful crises. ( 21527519 )
- Nonetheless, PAH drugs can be considered on an individual basis, for a subset of patients with precapillary pulmonary hypertension. (Murray 2022)
To keep this page small and fast, questions & discussion about this post can be found on another page here .
Guide to emoji hyperlinks
- 📄 = Link to open-access journal article.
- 23809320 Mikhalkova D, Fenstad ER, Miller WL. 34-year-old man with exertional syncope, dyspnea, and chest pain. Mayo Clin Proc. 2013 Jul;88(7):756-60. doi: 10.1016/j.mayocp.2012.09.012 [ PubMed ]
- 28991555 Chaddha U, Puscas I, Prosper A, Ganesh S, Yaghmour B. A 63-Year-Old Woman With Neurofibromatosis Type 1 and Pulmonary Hypertension With Worsening Hypoxemia. Chest. 2017 Oct;152(4):e89-e93. doi: 10.1016/j.chest.2017.05.014 [ PubMed ]
- 30526986 Rodriguez-Andoney JJ, Jimenez-Zamora V, Rivero-Sigarroa E, Hernandez-Oropeza JL, García-Juárez I, Dominguez-Cherit G. A 44-Year-Old Woman With Sudden Breathlessness, Tightness in Chest, and Hypotension After Extubation in the Early Postoperative Period After Liver Transplantation. Chest. 2018 Dec;154(6):e177-e180. doi: 10.1016/j.chest.2018.07.004 [ PubMed ]
- 31174660 Jalil BA, Smith JS, El-Kersh K. A 34-Year-Old Woman With Hoarseness of Voice and an Abnormal Echocardiogram. Chest. 2019 Jun;155(6):e163-e166. doi: 10.1016/j.chest.2019.01.021 [ PubMed ]
- 32654737 Walia A, Singh I, Ryu C, Lutchmansingh DD. A 50-Year-Old Woman With Limited Scleroderma Presenting With Shortness of Breath. Chest. 2020 Jul;158(1):e37-e40. doi: 10.1016/j.chest.2020.02.041 [ PubMed ]
- 32768078 Duvvuri PD, Liu J, Bhardwaj C. A 59-Year-Old Woman With Shortness of Breath and Chest Pain. Chest. 2020 Aug;158(2):e65-e69. doi: 10.1016/j.chest.2020.02.059 [ PubMed ]
- 34022029 Homsy E, Smith S. A 26-Year-Old Woman With Dyspnea on Exertion. Chest. 2021 Apr;159(4):e257-e260. doi: 10.1016/j.chest.2020.10.044 [ PubMed ]
- 34295399 Cullivan S, Morris J, McCormack C, Alameeri A, Gaine SP, McCullagh B. An interesting case of progressive dyspnoea and diffuse mediastinal adenopathy in a 25-year-old man. Breathe (Sheff). 2021 Mar;17(1):200289. doi: 10.1183/20734735.0289-2020 [ PubMed ]
- 34366049 Rogers E, Moffet EW, Allen N, Rivera-Zengotita M, Harden C, Ataya A. A 60-Year-Old Man With Dyspnea, Proximal Muscle Weakness, and Pulmonary Arterial Hypertension. Chest. 2021 Aug;160(2):e225-e231. doi: 10.1016/j.chest.2021.03.040 [ PubMed ]
- 34743853 Fakili F, Duzen IV, Kaplan M, Bayram NG. A 24-Year-Old Woman With Dyspnea, Chest Pain, and Dry Cough. Chest. 2021 Nov;160(5):e503-e506. doi: 10.1016/j.chest.2021.05.064 [ PubMed ]
- 36017548 Humbert M, Kovacs G, Hoeper MM, Badagliacca R, Berger RMF, Brida M, Carlsen J, Coats AJS, Escribano-Subias P, Ferrari P, Ferreira DS, Ghofrani HA, Giannakoulas G, Kiely DG, Mayer E, Meszaros G, Nagavci B, Olsson KM, Pepke-Zaba J, Quint JK, Rådegran G, Simonneau G, Sitbon O, Tonia T, Toshner M, Vachiery JL, Vonk Noordegraaf A, Delcroix M, Rosenkranz S; ESC/ERS Scientific Document Group. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J. 2022 Oct 11;43(38):3618-3731. doi: 10.1093/eurheartj/ehac237 [ PubMed ]
- 36813291 Cullivan S, Gaine S, Sitbon O. New trends in pulmonary hypertension. Eur Respir Rev. 2023 Feb 21;32(167):220211. doi: 10.1183/16000617.0211-2022 [ PubMed ]
- 37026538 Maron BA. Revised Definition of Pulmonary Hypertension and Approach to Management: A Clinical Primer. J Am Heart Assoc. 2023 Apr 18;12(8):e029024. doi: 10.1161/JAHA.122.029024 [ PubMed ]
- 37775164 Virsinskaite R, Karia N, Kotecha T, Schreiber BE, Coghlan JG, Knight DS. Pulmonary hypertension – the latest updates for physicians. Clin Med (Lond). 2023 Sep;23(5):449-454. doi: 10.7861/clinmed.2023-23.5.Cardio4 [ PubMed ]
- Shah , P. L., Herth, F. J., Lee, G., & Criner, G. J. (2018). Essentials of Clinical pulmonology. In CRC Press eBooks. https://doi.org/10.1201/9781315113807
- Shepard, JO . (2019). Thoracic Imaging The Requisites (Requisites in Radiology) (3rd ed.). Elsevier.
- Walker C & Chung JH (2019). Muller’s Imaging of the Chest: Expert Radiology Series . Elsevier.
- Palange, P., & Rohde, G. (2019). ERS Handbook of Respiratory Medicine. European Respiratory Society.
- Rosado-De-Christenson , M. L., Facr, M. L. R. M., & Martínez-Jiménez, S. (2021). Diagnostic imaging: chest. Elsevier.
- Murray & Nadel : Broaddus, V. C., Ernst, J. D., MD, King, T. E., Jr, Lazarus, S. C., Sarmiento, K. F., Schnapp, L. M., Stapleton, R. D., & Gotway, M. B. (2021). Murray & Nadel’s Textbook of Respiratory Medicine, 2-Volume set. Elsevier.
- Fishman's : Grippi, M., Antin-Ozerkis, D. E., Cruz, C. D. S., Kotloff, R., Kotton, C. N., & Pack, A. (2023). Fishman’s Pulmonary Diseases and Disorders, Sixth Edition (6th ed.). McGraw Hill / Medical.
The Internet Book of Critical Care is an online textbook written by Josh Farkas ( @PulmCrit ), an associate professor of Pulmonary and Critical Care Medicine at the University of Vermont.
We are the EMCrit Project , a team of independent medical bloggers and podcasters joined together by our common love of cutting-edge care, iconoclastic ramblings, and FOAM.
Resus Leadership Academy

Subscribe by Email
Insert/edit link.
Enter the destination URL
Or link to existing content
Learn how UpToDate can help you.
Select the option that best describes you
- Medical Professional
- Resident, Fellow, or Student
- Hospital or Institution
- Group Practice
- Patient or Caregiver
- Find in topic
RELATED TOPICS
INTRODUCTION
The clinical features, diagnostic evaluation, and diagnostic criteria for PH are reviewed here. Epidemiology, pathogenesis, treatment, and prognosis are discussed separately. (See "The epidemiology and pathogenesis of pulmonary arterial hypertension (Group 1)" and "Treatment of pulmonary arterial hypertension (group 1) in adults: Pulmonary hypertension-specific therapy" and "Treatment and prognosis of pulmonary arterial hypertension in adults (group 1)" .)
ETIOLOGIES AND TERMINOLOGY
PH can also be classified as pre- or postcapillary PH. Precapillary PH is due to a primary elevation of pressure in the PA system alone (eg, PAH), while postcapillary PH is due to elevations of pressure in the pulmonary venous and pulmonary capillary systems (pulmonary venous hypertension; eg, group 2). In practice, some patients have mixed pre- and postcapillary features ( table 2 ).
CLINICAL MANIFESTATIONS
- Patient Care & Health Information
- Diseases & Conditions
- Pulmonary hypertension
Pulmonary hypertension is hard to diagnose early because it's not often found during a routine physical exam. Even when pulmonary hypertension is more advanced, its symptoms are similar to those of other heart and lung conditions.
To diagnose pulmonary hypertension, a health care professional examines you and asks about your symptoms. You'll likely be asked questions about your medical and family history.
Tests done to help diagnose pulmonary hypertension may include:
- Blood tests. Blood tests can help find the cause of pulmonary hypertension or show signs of complications.
- Chest X-ray. A chest X-ray creates pictures of the heart, lungs and chest. It may be used to check for other lung conditions that can cause pulmonary hypertension.
- Electrocardiogram (ECG or EKG). This simple test records the electrical activity of the heart. It can show changes in the heartbeat.
Echocardiogram. Sound waves are used to create moving images of the beating heart. An echocardiogram shows blood flow through the heart. This test may be done to help diagnose pulmonary hypertension or to determine how well treatments are working.
Sometimes, an echocardiogram is done while exercising on a stationary bike or treadmill to learn how activity affects the heart. If you have this test, you may be asked to wear a mask that checks how well the heart and lungs use oxygen and carbon dioxide.
Right heart catheterization. If an echocardiogram shows pulmonary hypertension, this test may be done to confirm the diagnosis.
During this procedure, a cardiologist places a thin, flexible tube called a catheter into a blood vessel, usually in the neck. The catheter is gently guided into the lower right heart chamber and the pulmonary artery. A doctor can then measure blood pressure in the main pulmonary arteries and the right ventricle.
Other tests may be done to check the condition of the lungs and pulmonary arteries. The following tests may give more information about the cause of pulmonary hypertension:
- Exercise stress tests. These tests often involve walking on a treadmill or riding a stationary bike while the heartbeat is watched. They can show how the heart reacts to exercise.
Computerized tomography (CT) scan. This test uses X-rays to create cross-sectional images of specific parts of the body. Dye called contrast may be given into a vein to help the blood vessels show up more clearly on the images.
A heart CT scan, called a cardiac CT scan, can show the size of the heart and any blockages in the pulmonary arteries. It can help diagnose lung diseases that might lead to pulmonary hypertension such as COPD or pulmonary fibrosis.
- Magnetic resonance imaging (MRI). This test uses magnetic fields and radio waves to create detailed images of the heart. It can show blood flow in the pulmonary arteries and determine how well the right lower heart chamber is working.
- Lung function test. For this test, you blow into a special device. The device measures how much air the lungs can hold. It shows how air flows in and out of the lungs.
- Sleep study. A sleep study measures brain activity, heart rate, blood pressure, oxygen levels and other things as you sleep. The test can help diagnose sleep apnea, which can cause pulmonary hypertension.
- Ventilation/perfusion (V/Q) scan. In this test, a radioactive tracer is given through a vein (IV). The tracer shows blood flow. You also may breathe in a tracer that shows airflow to the lungs. A V/Q scan can show whether blood clots are causing symptoms of pulmonary hypertension.
- Lung biopsy. Rarely, a sample of tissue may be taken from the lung to check for a possible cause of pulmonary hypertension.
- Genetic testing
Screening for gene changes that cause pulmonary hypertension may be recommended. If you have these gene changes, other family members may need to be screened too.
Pulmonary hypertension functional classification
Once a diagnosis of pulmonary hypertension is confirmed, the condition is classified according to how the symptoms affect you and your ability to do everyday tasks.
Pulmonary hypertension may fall into one of the following groups:
- Class I. Pulmonary hypertension is diagnosed, but there are no symptoms during rest or exercise.
- Class II. There are no symptoms at rest. Everyday chores or activities such as going to work or the grocery store may cause some shortness of breath or mild chest pain. There's a slight limitation of physical activity.
- Class III. It's comfortable at rest, but doing simple tasks such as bathing, dressing or preparing meals causes fatigue, shortness of breath and chest pain. The ability to do physical activity becomes very limited.
- Class IV. Symptoms occur at rest and during physical activity. Any type of activity causes increasing discomfort.
Your health care team may use a risk calculator that considers your symptoms and test results to understand what type of treatment is needed. This is called pulmonary hypertension risk stratification.
- Care at Mayo Clinic
Our caring team of Mayo Clinic experts can help you with your pulmonary hypertension-related health concerns Start Here
More Information
Pulmonary hypertension care at Mayo Clinic
- Cardiac catheterization
- Chest X-rays
- Echocardiogram
There's no cure for pulmonary hypertension. But treatment is available to improve symptoms and prolong life, and to keep the disease from getting worse. You also may get treatments for any health problem that might be causing pulmonary hypertension.
It often takes some time to find the most appropriate treatment for pulmonary hypertension. The treatments are often complex. You usually need a lot of health checkups.
Medications
If you have pulmonary hypertension, you may get medicines to treat your symptoms and help you feel better. Medicines also may be used to treat or prevent complications. Treatment may include:
Medicines to relax blood vessels. Also called vasodilators, these medicines help open narrowed blood vessels and improve blood flow. The medicine comes in many forms. It may be breathed in, taken by mouth or given by IV . Some types are given continuously through a small pump attached to the body.
Examples of vasodilators to treat pulmonary hypertension include epoprostenol (Flolan, Veletr), treprostinil (Remodulin, Tyvaso, others), Iloprost (Ventavis) and selexipag (Uptravi).
- Soluble guanylate cyclase (sGC) stimulators. This type of medicine relaxes the pulmonary arteries and lowers pressure in the lungs. Examples include riociguat (Adempas). Do not take these medicines if you're pregnant.
- Medicines to widen blood vessels. Medicines called endothelin receptor antagonists reverse the effect of a substance in the walls of blood vessels that causes them to narrow. Such medicines include bosentan (Tracleer), macitentan (Opsumit) and ambrisentan (Letairis). They may improve energy level and symptoms. Do not take these medicines if you are pregnant.
- Medicines to increase blood flow. Medicines called phosphodiesterase 5 (PDE5) inhibitors may be used to increase blood flow through the lungs. These medicines also are used to treat erectile dysfunction. They include sildenafil (Revatio, Viagra) and tadalafil (Adcirca, Alyq, Cialis).
- High-dose calcium channel blockers. These medicines help relax the muscles in the walls of blood vessels. They include amlodipine (Norvasc), diltiazem (Cardizem, Tiazac, others) and nifedipine (Procardia). Although calcium channel blockers can be effective, only a small number of people with pulmonary hypertension improve while taking them.
- Blood thinners. Also called anticoagulants, these medicines help prevent blood clots. One example is warfarin (Jantoven). Blood-thinning medicines slow the clotting process. The medicines can increase the risk of bleeding. This is especially true if you're having surgery or a procedure that enters the body or creates an opening in the skin. Talk to your health care team about your risk.
- Digoxin (Lanoxin). This medicine helps the heart beat stronger and pump more blood. It can help control irregular heartbeats.
- Water pills, also called diuretics. These medicines help the kidneys remove excess fluid from the body. This reduces the amount of work the heart has to do. Diuretics also may be used to reduce fluid buildup in the lungs, legs and belly area.
- Oxygen therapy. Breathing pure oxygen is sometimes recommended as a treatment for pulmonary hypertension. This treatment may be suggested if you live at a high altitude or have sleep apnea. Some people with pulmonary hypertension need oxygen therapy all the time.
Surgery or other procedures
If medicines do not help control the symptoms of pulmonary hypertension, surgery may be recommended. Surgeries and procedures to treat pulmonary hypertension may include:
- Atrial septostomy. This treatment may be recommended if medicines don't control pulmonary hypertension symptoms. In an atrial septostomy, a doctor creates an opening between the upper left and right chambers of the heart. The opening reduces the pressure on the right side of the heart. Potential complications include irregular heartbeats called arrhythmias.
- Lung or heart-lung transplant. Sometimes, a lung or heart-lung transplant may be needed, especially for younger people who have idiopathic pulmonary arterial hypertension. After a transplant, medicine must be taken for life to help reduce the chance of rejection.
- Extracorporeal membrane oxygenation (ECMO)
- Lung transplant
Clinical trials
Explore Mayo Clinic studies testing new treatments, interventions and tests as a means to prevent, detect, treat or manage this condition.
Lifestyle and home remedies
Lifestyle changes may help improve pulmonary hypertension symptoms. Try these tips:
- Eat healthy. Eat a healthy diet rich in whole grains, fruits and vegetables, lean meats, and low-fat dairy products. Try to stay away from saturated fat, trans fat and cholesterol. Limit salt.
- Stay as active as possible and manage weight. Even mild forms activity might be too exhausting for some people who have pulmonary hypertension. For others, moderate exercise, such as walking, might be helpful — especially when done during oxygen therapy. Your health care team can help you plan an appropriate exercise program.
- Don't smoke. If you smoke, the most important thing you can do for your heart and lungs is to stop. If you need support quitting, ask your health care team for treatment that can help. Avoid secondhand smoke too, if possible.
- Get plenty of rest. Resting can reduce tiredness related to pulmonary hypertension.
- Avoid high altitudes. High altitudes can make pulmonary hypertension worse. If you live at an altitude of 8,000 feet (2,438 meters) or higher, you might be told to consider moving to a lower altitude.
- Avoid activities that can excessively lower blood pressure. These include sitting in a hot tub or sauna or taking long hot baths or showers. Such activities lower blood pressure and can cause fainting or even death. Also, do not do activities that cause a lot of straining, such as lifting heavy objects or weights.
- Give your health care team a list of your medicines. Some medicines can make pulmonary hypertension worse or affect its treatment.
- Get regular health checkups. Tell your health care team about any new or worsening symptoms or medicine side effects. If pulmonary hypertension affects your quality of life, ask about options that could help.
- Get recommended vaccines. Respiratory infections can cause serious health concerns for people with pulmonary hypertension. Ask your health care team about recommend vaccines to prevent common viral infections.
- Talk to a health care professional before becoming pregnant. Pulmonary hypertension can cause serious complications to both mother and baby during pregnancy. Birth control pills can increase the risk of blood clots. Talk to your health care team about other birth control options.
Coping and support
Connecting with others who are going through similar situations may help you ease and manage stress. Ask your health care team if there are any pulmonary hypertension support groups in your area.
Preparing for your appointment
If you think that you might have or be at risk of pulmonary hypertension, make an appointment for a health checkup.
There's often a lot to discuss at your appointment, so it's a good idea to be prepared. Here's some information to help you get ready for your appointment.
What you can do
- Be aware of any pre-appointment restrictions. When you make your appointment, ask if there is anything you need to do before your checkup. For example, you might be told not to eat or drink before some medical tests.
- Write down any symptoms you're having, including any that might seem unrelated to pulmonary hypertension. Try to remember when they began. Be specific, such as days, weeks and months.
- Make a list of important personal information, including any family history of pulmonary hypertension, lung disease, heart disease, stroke, high blood pressure or diabetes, and any major stresses or recent life changes.
- Make a list of all medicines, as well as any vitamins, herbal products or other supplements that you're taking.
- Take a family member or friend along, if possible. Someone who goes with you can help you remember information you're given.
- Be prepared to discuss your diet and exercise habits. If you don't already follow a diet or exercise routine, talk to your health care team about any challenges you might face in getting started.
- Make a list of questions to ask your health care team. List your questions from most important to least important in case time runs out.
For pulmonary hypertension, some basic questions to ask your health care team are:
- What is likely causing my symptoms or condition?
- What are other possible causes for my symptoms or condition?
- What kinds of tests do I need?
- What's the most appropriate treatment?
- Is there a generic alternative to the medicine you're prescribing?
- What are the options to the primary treatment that you're suggesting?
- What's an appropriate level of physical activity?
- How often should I be checked for changes in my condition?
- I have other health conditions. How can I best manage them together?
- Are there any restrictions that I need to follow?
- Should I see a specialist?
- Are there any brochures or other printed material that I can have? What websites do you suggest?
Don't hesitate to ask other questions.
What to expect from your doctor
Your doctor and other members of your health care team may ask you many questions. Being ready to answer them might give you more time to discuss any concerns. You may be asked:
- When did you first begin having symptoms?
- Do you always have symptoms, or do they come and go?
- How severe are the symptoms?
- What, if anything, seems to improve symptoms?
- What, if anything, seems to make symptoms worse?
What you can do in the meantime
It's never too late to make healthy lifestyle changes, such as quitting smoking, reducing salt and eating a healthy diet. These changes may help prevent pulmonary hypertension from getting worse.
Living with pulmonary hypertension?
Connect with others like you for support and answers to your questions in the Transplants support group on Mayo Clinic Connect, a patient community.
Transplants Discussions

74 Replies Tue, May 14, 2024

1670 Replies Tue, May 14, 2024

377 Replies Mon, May 13, 2024
- Pulmonary hypertension — High blood pressure in the heart-to-lung system. American Heart Association. https://www.heart.org/en/health-topics/high-blood-pressure/the-facts-about-high-blood-pressure/pulmonary-hypertension-high-blood-pressure-in-the-heart-to-lung-system. Accessed May 10, 2023.
- Pulmonary hypertension. National Heart, Lung, and Blood Institute. https://www.nhlbi.nih.gov/health/pulmonary-hypertension. Accessed May 10, 2023.
- Klinger JR, et al. Therapy for pulmonary arterial hypertension in adults: Update of the CHEST guideline and expert panel report. Chest. 2019; doi:10.1016/j.chest.2018.11.030.
- AskMayoExpert. Pulmonary hypertension (adult). Mayo Clinic; 2022.
- Rubin LJ, et al. Clinical features and diagnosis of pulmonary hypertension of unclear etiology in adults. https://www.uptodate.com/contents/search. Accessed Nov. 19, 2022.
- Hopkins W, et al. Treatment of pulmonary arterial hypertension (group 1) in adults: Pulmonary hypertension-specific therapy. https://www.uptodate.com/contents/search. Accessed Nov. 19, 2022.
- Fuster V, et al., eds. Pulmonary hypertension. In: Fuster and Hurst's the Heart. 15th ed. McGraw Hill; 2022. https://accessmedicine.mhmedical.com. Accessed Nov. 19, 2022.
- Ami TR. Allscripts EPSi. Mayo Clinic. Dec. 21, 2022.
- Ferri FF. Pulmonary hypertension. In: Ferri's Clinical Advisor 2023. Elsevier; 2023. https://www.clinicalkey.com. Accessed Nov. 19, 2022.
- Connolly HM. Evaluation and prognosis of Eisenmenger syndrome. https://www.uptodate.com/contents/search. Accessed Nov. 19, 2022.
- Olson EJ (expert opinion). Mayo Clinic. Jan. 24, 2023.
- Simonneau G, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. The European Respiratory Journal. 2019; doi:10.1183/13993003.01913-2018.
- Gelzinis TA. Pulmonary hypertension in 2021: Part I — definition, classification, pathophysiology, and presentation. Journal of Cardiothoracic and Vascular Anesthesia. 2021; doi:10.1053/j.jvca.2021.06.036.
- Humbert M, et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. European Heart Journal. 2022; doi:10.1093/eurheartj/ehac237.
- Highland KB, et al. Development of the pulmonary hypertension functional classification self‑report: A patient version adapted from the World Health Organization functional classification measure. Health and Quality of Life Outcomes. 2021; doi:10.1186/s12955-021-01782-0.
- Mankad R (expert opinion). Mayo Clinic. Feb. 7, 2023.
- Frantz RP (expert opinion). Mayo Clinic. May 3, 2023.
- ED Drugs for Pulmonary Hypertension
Associated Procedures
Products & services.
- A Book: Mayo Clinic Family Health Book, 5th Edition
- Assortment of Compression Products at Mayo Clinic Store
- Newsletter: Mayo Clinic Health Letter — Digital Edition
Mayo Clinic in Rochester, Minnesota, has been recognized as one of the top Cardiology & Heart Surgery hospitals in the nation for 2023-2024 by U.S. News & World Report.
- Symptoms & causes
- Diagnosis & treatment
- Doctors & departments
Mayo Clinic does not endorse companies or products. Advertising revenue supports our not-for-profit mission.
- Opportunities
Mayo Clinic Press
Check out these best-sellers and special offers on books and newsletters from Mayo Clinic Press .
- Mayo Clinic on Incontinence - Mayo Clinic Press Mayo Clinic on Incontinence
- The Essential Diabetes Book - Mayo Clinic Press The Essential Diabetes Book
- Mayo Clinic on Hearing and Balance - Mayo Clinic Press Mayo Clinic on Hearing and Balance
- FREE Mayo Clinic Diet Assessment - Mayo Clinic Press FREE Mayo Clinic Diet Assessment
- Mayo Clinic Health Letter - FREE book - Mayo Clinic Press Mayo Clinic Health Letter - FREE book
Your gift holds great power – donate today!
Make your tax-deductible gift and be a part of the cutting-edge research and care that's changing medicine.
Pulmonary Hypertension
- Pathophysiology |
- Symptoms and Signs |
- Diagnosis |
- Prognosis |
- Treatment |
- Key Points |
Pulmonary hypertension is increased pressure in the pulmonary circulation. It has many secondary causes; some cases are idiopathic. In pulmonary hypertension, pulmonary vessels may become constricted, pruned, lost, and/or obstructed. Severe pulmonary hypertension leads to right ventricular overload and failure. Symptoms are fatigue, exertional dyspnea, and, occasionally, chest discomfort and syncope. Diagnosis is made by finding elevated pulmonary artery pressure (estimated by echocardiography and confirmed by right heart catheterization). Treatment is with pulmonary vasodilators and diuretics. In some advanced cases, lung transplantation is an option. Prognosis is poor overall if a treatable secondary cause is not found.
There are three distinct hemodynamic profiles of pulmonary hypertension (see also table Hemodynamic Profiles of Pulmonary Hypertension ):
Pre-capillary pulmonary hypertension
Post-capillary pulmonary hypertension
Combined pre- and post-capillary pulmonary hypertension
Etiology of Pulmonary Hypertension
Many conditions and drugs cause pulmonary hypertension. The most common overall causes of pulmonary hypertension are
Left heart failure , including diastolic dysfunction
Parenchymal lung disease with hypoxia
Several other causes of pulmonary hypertension include sleep apnea , connective tissue disorders , and recurrent pulmonary embolism .
Pulmonary hypertension is currently classified into 5 groups (see table Classification of Pulmonary Hypertension ) based on a number of pathologic, physiologic, and clinical factors. In the first group (pulmonary arterial hypertension [PAH]), the primary disorder affects the small pulmonary arterioles.
A small number of cases of AH occur sporadically, unrelated to any identifiable disorder; these cases are termed idiopathic PAH.
Hereditary forms of PAH (autosomal dominant with incomplete penetrance) have been identified; mutations of the following genes have been found:
Activin-like kinase type 1 receptor ( ALK-1 )
Bone morphogenetic protein receptor type 2 ( BMPR2 )
Caveolin 1 ( CAV1 )
Endoglin ( ENG )
Growth differentiation factor 2 ( GDF2 )
Potassium channel subfamily K member 3 ( KCNK3 )
Mothers against decapentaplegic homologue 9 ( SMAD9 )
T-box transcription factor 4 ( TBX4 )
Mutations in BMPR2 cause 75% of cases. The other mutations are much less common, occurring in about 1% of cases.
In about 20% of cases of hereditary PAH, the causative mutations are unidentified.
A mutation in the eukaryotic translation initiation factor 2 alpha kinase 4 gene ( EIF2AK4 ) has been linked to pulmonary veno-occlusive disease, a form of PAH Group 1' ( 1, 2 ).
Certain drugs and toxins 3 ). Selective serotonin reuptake inhibitors taken by pregnant women are a risk for development of persistent pulmonary hypertension of the newborn 4 ).
Patients with hereditary causes of hemolytic anemia , such as sickle cell disease , are at high risk of developing pulmonary hypertension (10% of cases based on right heart catheterization criteria). The mechanism is related to intravascular hemolysis and release of cell-free hemoglobin into the plasma, which scavenges nitric oxide, generates reactive oxygen species, and activates the hemostatic system. Other risk factors for pulmonary hypertension in sickle cell disease include iron overload , liver dysfunction, thrombotic disorders , and chronic kidney disease .
Etiology references
1. Eyries M, Montani D, Girerd B, et al : EIF2AK4 mutations cause pulmonary veno-occlusive disease, a recessive form of pulmonary hypertension. Nat Genet 46(1):65-9, 2014. doi: 10.1038/ng.2844
2. Girerd B, Weatherald J, Montani D, Humbert M : Heritable pulmonary hypertension: from bench to bedside. Eur Respir Rev 26(145):170037, 2017. doi: 10.1183/16000617.0037-2017
3. Cornet L, Khouri C, Roustit M, et al : Pulmonary arterial hypertension associated with protein kinase inhibitors: a pharmacovigilance-pharmacodynamic study. Eur Respir J 9;53(5):1802472, 2019. doi: 10.1183/13993003.02472-2018
4. Simonneau G, Montani D, Celermajer DS, et al : Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J 53(1):1801913, 2019. doi: 10.1183/13993003.01913-2018
Pathophysiology of Pulmonary Hypertension
Pathophysiologic mechanisms that cause pulmonary hypertension include
Increased pulmonary vascular resistance
Increased pulmonary venous pressure
Increased pulmonary venous flow due to congenital heart diseases
Increased pulmonary vascular resistance BMPR2 gene account for most cases of hereditary PAH and also occurs in idiopathic PAH. Aberrant BMPR2 signaling disturbs the TGF-β/BMP balance, favoring a pro-proliferative and anti-apoptotic response in pulmonary artery smooth muscle and endothelial cells. BMPR2 signaling, therefore, has become an increasingly studied drug target for pulmonary hypertension.
Increased pulmonary venous pressure is typically caused by disorders that affect the left side of the heart and raise left chamber pressures, which ultimately lead to elevated pressure in the pulmonary veins. Elevated pulmonary venous pressures can cause acute damage to the alveolar-capillary wall and subsequent edema. Persistently high pressures may eventually lead to irreversible thickening of the walls of the alveolar-capillary membrane, decreasing lung diffusion capacity. The most common setting for pulmonary venous hypertension is in left heart failure with preserved ejection fraction (HFpEF), typically in older women who have hypertension and metabolic syndrome .
In pulmonary hypertension secondary to HFpEF, certain hemodynamic parameters predict an increased risk of death. These parameters include
Transpulmonary gradient (TPG, defined as the mean pulmonary artery pressure to pulmonary artery occlusion pressure gradient) > 12 mm Hg
Pulmonary vascular resistance (PVR, defined as the TPG divided by the cardiac output) ≥ 3 Woods units
Diastolic pulmonary gradient (DPG, defined as the pulmonary artery diastolic pressure to pulmonary artery occlusion pressure gradient) ≥ 7 mm Hg
In most patients, pulmonary hypertension eventually leads to right ventricular hypertrophy followed by dilation and right ventricular failure . Right ventricular failure limits cardiac output during exertion.
Increased pulmonary venous blood flow due to congenital heart disease can cause pulmonary hypertension. This can occur in conditions such as atrial septal defects , ventricular septal defects , and patent ductus arteriosus , presumably through the development of characteristic pulmonary vascular lesions. However, the true effect of increased pulmonary blood flow is poorly defined and increased flow may lead to vascular obstruction only with concomitant pulmonary vascular resistance or a second stimulus.
Symptoms and Signs of Pulmonary Hypertension
Progressive exertional dyspnea and easy fatigability occur in almost all patients. Atypical chest discomfort and exertional light-headedness or presyncope may accompany dyspnea and indicate more severe disease. These symptoms are due primarily to insufficient cardiac output caused by right heart failure. Raynaud syndrome occurs in about 10% of patients with idiopathic pulmonary arterial hypertension; the majority are women. Hemoptysis is rare but may be fatal. Hoarseness due to recurrent laryngeal nerve compression by an enlarged pulmonary artery (ie, Ortner syndrome) also occurs rarely.
In advanced disease, signs of right heart failure may include right ventricular heave, widely split 2nd heart sound (S2), an accentuated pulmonic component (P2) of S2, a pulmonary ejection click, a right ventricular 3rd heart sound (S3), tricuspid regurgitation murmur, and jugular vein distention, possibly with v-waves. Liver congestion and peripheral edema are common late manifestations. Pulmonary auscultation is usually normal. Patients also may have manifestations of causative or associated disorders.
Diagnosis of Pulmonary Hypertension
Exertional dyspnea
Initial confirmation: Chest x-ray, ECG, and echocardiography
Identification of underlying disorder: Spirometry, ventilation/perfusion scanning or CT angiography, high-resolution CT (HRCT) of the chest, pulmonary function testing, polysomnography, HIV testing, complete blood count, liver tests, and autoantibody testing
Confirmation of the diagnosis and gauging severity: Pulmonary artery (right heart) catheterization
Additional studies to determine severity: 6-minute walk distance and plasma levels of N-terminal pro-brain natriuretic peptide (NT-proBNP) or BNP
Pulmonary hypertension is suspected in patients with significant exertional dyspnea who are otherwise relatively healthy and have no history or signs of other disorders known to cause pulmonary symptoms.
Patients initially undergo chest x-ray , spirometry , and ECG to identify more common causes of dyspnea, followed by transthoracic Doppler echocardiography to assess right ventricular function and pulmonary artery systolic pressures as well as to detect structural left heart disease that might be causing pulmonary hypertension. Complete blood count is obtained to document the presence or absence of erythrocytosis, anemia, and thrombocytopenia.
The most common x-ray finding in pulmonary hypertension is enlarged hilar vessels that rapidly prune into the periphery and a right ventricle that fills the anterior airspace on lateral view. Spirometry and lung volumes may be normal or show mild restriction, and diffusing capacity for carbon monoxide (DLCO) is usually reduced. Other ECG findings include right axis deviation, R > S in V1, S1Q3T3 (suggesting right ventricular hypertrophy), and peaked P waves (suggesting right atrial dilation) in lead II.
Additional tests are obtained as indicated to diagnose secondary causes that are not apparent clinically. These tests can include
Ventilation/perfusion scanning or CT angiography to detect thromboembolic disease
HRCT for detailed information about lung parenchymal disorders in patients in whom CT angiography is not done
Pulmonary function tests to identify obstructive or restrictive lung disease
Serum autoantibody tests (eg, antinuclear antibodies [ANA], rheumatoid factor [RF], Scl-70 [topoisomerase I], anti-Ro (anti-SSA), anti-ribonucleoprotein [anti-RNP], and anticentromere antibodies) to gather evidence for or against associated autoimmune disorders
Chronic thromboembolic pulmonary hypertension is suggested by CT angiography or ventilation/perfusion (VQ) scan findings and is confirmed by arteriography. CT angiography is useful to evaluate proximal clot and fibrotic encroachment of the vascular lumen. Other tests, such as HIV testing, liver tests, and polysomnography, are done in the appropriate clinical context.
When the initial evaluation suggests a diagnosis of pulmonary hypertension, pulmonary artery catheterization is necessary to measure the following:
Right atrial pressure
Right ventricular pressure
Pulmonary artery pressure
Pulmonary artery occlusion pressure
Cardiac output
Left ventricular diastolic pressure
Right-sided oxygen saturation should be measured to exclude left-to-right shunt through atrial septal defect . Although finding a mean pulmonary arterial pressure of > 20 mm Hg and a pulmonary artery occlusion pressure ≤ 15 mm Hg in the absence of an underlying disorder identifies pulmonary arterial hypertension (PAH), most patients with PAH present with substantially higher pressure (eg, mean of 60 mm Hg).
adenosine , are often given during catheterization. Decreasing right-sided pressures in response to these drugs may help in the choice of drugs for treatment. Lung biopsy, once widely done, is neither needed nor recommended because of its associated high morbidity and mortality.
Echocardiography findings of right heart systolic dysfunction (eg, tricuspid annular plane systolic excursion) and certain right heart catheterization results (eg, low cardiac output, very high mean pulmonary artery pressures, and high right atrial pressures) indicate that pulmonary hypertension is severe.
Other indicators of severity in pulmonary hypertension are assessed to evaluate prognosis and to help monitor responses to therapy. They include a low 6-minute walk distance and high plasma levels of N-terminal pro-brain natriuretic peptide (NT-pro-BNP) or brain natriuretic peptide (BNP).
Once pulmonary hypertension is diagnosed, the patient's family history should be reviewed to detect possible genetic transmission (eg, premature deaths in otherwise healthy members of the extended family). In familial PAH, genetic counseling is needed to advise mutation carriers of the risk of disease (about 20%) and to advocate serial screening with echocardiography. Testing for mutations in the BMPR2 gene in idiopathic PAH can help identify family members at risk. If patients are negative for BMPR2 , gene testing for SMAD9 , KCN3 , and CAV1 can further help identify family members at risk.
Prognosis for Pulmonary Hypertension
Five-year survival for treated patients is about 50%. However, some patient registries suggest lower mortality (eg, 20 to 30% at 3 to 5 years in the French registry and 10 to 30% at 1 to 3 years in the REVEAL registry), presumably because currently available treatments are superior. Indicators of a poorer prognosis include
Lack of response to vasodilators
Reduced overall physical functioning
Low 6-minute walk distance
High plasma levels of NT-pro-BNP or BNP
Echocardiographic indicators of right heart systolic dysfunction (eg, a tricuspid annular plane systolic excursion of < 1.6 cm, dilated right ventricle, flattened interventricular septum with paradoxical septal motion, and pericardial effusion)
Right heart catheterization showing low cardiac output, very high mean pulmonary artery pressures, and/or high right atrial pressures
Patients with systemic sclerosis, sickle cell disease, or HIV infection with pulmonary arterial hypertension (PAH) have a worse prognosis than those without PAH. For example, patients with sickle cell disease and PAH have a 40% 4-year mortality rate.
Treatment of Pulmonary Hypertension
Avoidance of activities that may exacerbate the condition (eg, cigarette smoking, high altitude, pregnancy, use of sympathomimetics)
Secondary pulmonary arterial hypertension: Treatment of the underlying disorder
Lung transplantation
Adjunctive therapy: Supplemental oxygen, diuretics, and/or anticoagulants
Pulmonary arterial hypertension, group 1
Treatment of pulmonary arterial hypertension (PAH) is rapidly evolving. Drugs target 4 aberrant pathways implicated in the development of PAH:
Endothelin pathway
Nitric oxide pathway
Prostacyclin pathway
BMPR2 pathway
The endothelin pathway
The nitric oxide pathway
The prostacyclin pathway 1 2 ).
The BMPR2 (bone morphogenic receptor type 2) pathway BMPR2 3 ).
Combination therapy 4
7, 8 9 10 ).
Guidelines for sequence of therapies may be evolving. The current recommendation is to do vasoactive testing in the catheterization laboratory. If patients are vasoreactive, they should be treated with a calcium channel blocker. Patients who are not vasoreactive should be treated based on their New York Heart Association class 11 ).
Selected subgroups are sometimes treated differently. Prostacyclin analogs, endothelin-receptor antagonists, and guanylate cyclase stimulators have been studied primarily in idiopathic PAH; however, these drugs can be used cautiously (attending to drug metabolism and drug-drug interactions) in patients with PAH due to connective tissue disease, HIV, or portopulmonary hypertension . Vasodilators should be avoided in patients with PAH due to pulmonary veno-occlusive disease due to the risk of catastrophic pulmonary edema ( 12 ).
Lung transplantation offers the only hope of cure but has high morbidity because of rejection (bronchiolitis obliterans syndrome) and infection. The 5-year survival rate is 50%. Lung transplantation is reserved for patients with NYHA class IV disease (defined as dyspnea associated with minimal activity, leading to bed to chair limitations) or complex congenital heart disease in whom all therapies have failed and who meet other health criteria to be a transplant candidate.
Adjunctive therapies
Pulmonary hypertension, groups 2 to 5
Primary treatment involves management of the underlying disorder. Patients with left-sided heart disease
Patients with lung disorders 13 14, 15 ).
The first-line treatment for patients with severe pulmonary hypertension secondary to chronic thromboembolic disease 5 16 17 ).
Patients with sickle cell disease
Treatment references
1. Barst RJ, Rubin LJ, Long WA, et al : A comparison of continuous intravenous epoprostenol ( prostacyclin ) with conventional therapy for primary pulmonary hypertension. N Engl J Med 334(5):296–301, 1996. doi: 10.1056/NEJM199602013340504
2. Sitbon O, Channick R, Chin KM, et al : Selexipag for the treatment of pulmonary arterial hypertension. N Engl J Med 373: 2522-33, 2015. doi: 10.1056/NEJMoa1503184
3. Humbert M, McLaughlin V, Gibbs JSR, et al : Sotatercept for the treatment of pulmonary arterial hypertension. N Engl J Med 384(13):1204–1215, 2021. doi: 10.1056/NEJMoa2024277
4. Galie N, Barbera JA, Frost AE, et al : Initial use of ambrisentan plus tadalafil in pulmonary arterial hypertension. N Engl J Med 373: 834-44, 2015. doi: 10.1056/NEJMoa1413687
5. Ghofrani HA, Galiè N, Grimminger F, et al : Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med 369(4): 330-40, 2013. doi: 10.1056/NEJMoa1209655
6. White RJ, Jerjes-Sanchez C, Bohns Meyer GM, et al : Combination therapy with oral treprostinil for pulmonary arterial hypertension. A double-blind placebo-controlled clinical trial. Am J Respir Crit Care Med 201(6):707–717, 2020. doi: 10.1164/rccm.201908-1640OC
7. Tamura Y, Channick RN : New paradigm for pulmonary arterial hypertension treatment. Curr Opin Pulm Med 22(5): 429-33, 2016. doi: 10.1097/MCP.0000000000000308
8. McLaughlin VV, Channick R, Chin K, et al : Effect of selexipag on morbidity/mortality in pulmonary arterial hypertension: Results of the GRIPHON study. J Am Coll Cardiol 65 (suppl): A1538, 2015.
9. Chin KM, Sitbon O, Doelberg M, et al : Three- versus two-drug therapy for patients with newly diagnosed pulmonary arterial hypertension. J Am Coll Cardiol 78(14):1393–1403, 2021. doi: 10.1016/j.jacc.2021.07.057
10. McLaughlin VV, Channick R, De Marco T, et al : Results of an expert consensus survey on the treatment of pulmonary arterial hypertension with oral prostacyclin pathway agents. Chest 157(4):955–965, 2020. doi: 10.1016/j.chest.2019.10.043
11. Condon DF, Nickel NP, Anderson R, et al : The 6th World Symposium on Pulmonary Hypertension: what's old is new. F1000Research 8:F1000 Faculty Rev-888, 2019. doi: 10.12688/f1000research.18811.1
12. Galiè N, Humbert M, Vachiery JL, et al : 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J 37(1): 67-119, 2016. doi: 10.1093/eurheartj/ehv317
13. Waxman A, Restrepo-Jaramillo R, Thenappan T, et al : Inhaled treprostinil in pulmonary hypertension due to interstitial lung disease. N Engl J Med 384(4):325–334, 2021. doi: 10.1056/NEJMoa2008470
14. Nathan SD, Behr J, Collard HR, et al : Riociguat for idiopathic interstitial pneumonia-associated pulmonary hypertension (RISE-IIP): a randomised, placebo-controlled phase 2b study. Lancet Respir Med 7(9):780–790, 2019. doi: 10.1016/S2213-2600(19)30250-4
15. Sahay S, Channick R, Chin K, et al : Macitentan in pulmonary hypertension (PH) due to chronic lung disease: Real-world evidence from OPUS/OrPHeUS. J Heart Lung Trans 40 (4) [Suppl]: S105-S106, 2021.
16. Ghofrani HA, Simonneau G, D'Armini AM, et al : Macitentan for the treatment of inoperable chronic thromboembolic pulmonary hypertension (MERIT-1): results from the multicentre, phase 2, randomised, double-blind, placebo-controlled study. Lancet Respir Med 5(10):785–794, 2017. doi:10.1016/S2213-2600(17)30305-3
17. Channick R, McLaughlin V, Chin K, et al : Treatment of chronic thromboembolic pulmonary hypertension (CTEPH): Real-world experience with macitentan . J Heart Lung Trans 38(4) [suppl]: S483, 2019. doi: https://www.jhltonline.org/article/S1053-2498(19)31231-8/fulltext
Pulmonary hypertension is classified into 5 groups.
Suspect pulmonary hypertension if patients have dyspnea unexplained by another clinically evident cardiac or pulmonary disorder.
Begin diagnostic testing with chest x-ray, spirometry, ECG, and transthoracic Doppler echocardiography.
Confirm the diagnosis by right heart catheterization.
Treat group 1 by giving combination therapy with vasodilators and, if these are ineffective, consider lung transplantation.
Treat group 4 with pulmonary thromboendarterectomy unless the patient is not a candidate for surgery.
Treat groups 2, 3, and 5 by managing the underlying disorder, treating symptoms, and sometimes using other measures.

- Cookie Preferences

Copyright © 2024 Merck & Co., Inc., Rahway, NJ, USA and its affiliates. All rights reserved.

An official website of the United States government
The .gov means it’s official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you’re on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
Preview improvements coming to the PMC website in October 2024. Learn More or Try it out now .
- Advanced Search
- Journal List
Pulmonary Arterial Hypertension: Pathophysiology and Treatment
Norris s. h. lan.
1 School of Medicine and Pharmacology, University of Western Australia, Perth 6009, Australia; ua.ude.awu.tneduts@15815112 (N.S.H.L.); ua.ude.awu.tneduts@70593112 (B.D.M.); ua.ude.awu.tneduts@85031112 (S.S.K.)
Benjamin D. Massam
Sandeep s. kulkarni, chim c. lang.
2 Division of Molecular and Clinical Medicine, Mailbox 2, Ninewells Hospital and Medical School, University of Dundee, Dundee DD1 9SY, UK
Pulmonary arterial hypertension (PAH), the first category of pulmonary hypertension, is a chronic and progressive disorder characterised by angioproliferative vasculopathy in the pulmonary arterioles, leading to endothelial and smooth muscle proliferation and dysfunction, inflammation and thrombosis. These changes increase pulmonary vascular resistance and subsequent pulmonary arterial pressure, causing right ventricular failure which leads to eventual death if untreated. The management of PAH has advanced rapidly in recent years due to improved understanding of the condition’s pathophysiology, specifically the nitric oxide, prostacyclin-thromboxane and endothelin-1 pathways. Five classes of drugs targeting these pathways are now available: phosphodiesterase-5 inhibitors, soluble guanylate cyclase stimulators, prostacyclin analogues, prostacyclin receptor agonists and endothelin receptor antagonists. These developments have led to substantial improvements in mortality rate in recent decades. Recently, long-term studies have demonstrated sustained progression-free survival and have created a new paradigm of initial combination therapy. Despite these targeted therapies, PAH is still associated with significant morbidity and mortality. As such, further research into broadening our understanding of PAH pathophysiology is underway with potential of increasing the repertoire of drugs available.
1. Introduction
Pulmonary hypertension (PH) consists of a group of diseases with a resting mean pulmonary artery pressure (mPAP) ≥ 25 mmHg as measured with right heart catheterization [ 1 ]. Since 1998, major revisions in the classification of PH have categorised the disease based on the anatomical site and aetiology. The latest revision, in 2013, is shown in Table 1 .
Classification of pulmonary hypertension [ 1 ].
BMPRII = bone morphogenetic protein receptor type II; ALK1 = activin receptor-like kinase 1; ENG = endoglin 1; SMAD9 = mothers against decapentaplegic homolog 9; CAV1 = caveolin-1; KCNK3 = Potassium channel subfamily K member 3; HIV = human immunodeficiency virus.
PH is considered a rare disease, with a population-based study in the Netherlands suggesting a prevalence rate of 2.6% based on echocardiographic findings [ 2 ]. According to one Australian cohort study, PH secondary to left heart disease was the subtype with the greatest prevalence but also conferred the highest mortality, followed by PH secondary to respiratory diseases [ 3 ].
Group 1 of PH is pulmonary arterial hypertension (PAH), which is defined as mPAP ≥ 25 mmHg, pulmonary artery wedge pressure (PAWP) ≤ 15 mmHg and pulmonary vascular resistance (PVR) > 3 Wood units [ 1 ]. PAH is further subcategorised according to its aetiology, with idiopathic PAH (iPAH) comprising the majority of cases, followed by PAH associated with connective tissue diseases (CTD) and congenital heart disease (see Table 1 ) [ 4 ]. In PAH, the pre-capillary arterioles are affected by an angioproliferative vasculopathy that increases the pulmonary vascular resistance, thereby increasing the right ventricular afterload with the resulting right heart failure being the ultimate cause of mortality [ 5 ]. When adequately treated, PAH exhibits the best prognosis when compared to other PH categories according to the aforementioned Australian study [ 3 ].
According to registries in the United Kingdom, the reported incidence and prevalence rates of PAH are 1.1–2.4 and 6.6–15.0 cases per million per year, respectively [ 6 ]. Contemporary data from the 2014 UK National Audit on Pulmonary Hypertension found that the median age of diagnosis of PAH was 60 years in females and 58 years in males, with more than 25% of patients being over 70 years—thereby refuting previous conceptions of PAH being a disease of the young [ 7 ]. Data from the United States-based registry to evaluate early and long-term pulmonary arterial hypertension disease management (REVEAL registry) demonstrated certain sex preponderances for specific subtypes of PAH, such as a greater female distribution in iPAH [ 8 ]. Males demonstrated a lower survival rate at both two and five years post-enrolment; however, subanalysis showed similar survival rates between women and men <60 years at enrolment but poorer survival rates in men >60 compared to women [ 8 ]. Overall median survival rates have improved dramatically over the past two decades (from 2.8 to 7 years in the aforementioned American registry), presumably due to a combination of significant advances in treatment strategies and patient support strategies [ 9 ].
2. Risk and Prognostic Factors of PAH
Drugs, toxins, CTD and certain infections (such as HIV and schistosomiasis) have been strongly associated with PAH, and have thus appeared as subcategories in the classification [ 10 , 11 ]. The drugs and toxins considered as definite risk factors include aminorex, fenfluramine, dexfenfluramine and selective serotonin reuptake inhibitors (SSRIs) [ 10 ]. In particular, iPAH has been found to be strongly associated with female gender, family history and genetic variants, especially bone morphogenetic protein receptor type 2 ( BMPRII ) mutations [ 4 , 12 ].
Prognostic stratification of PAH in clinical practice requires extensive assessment and investigation at specialised PH centres. As highlighted in Table 2 , the 2015 ESC/ERS (European Society of Cardiology and the European Respiratory Society) guidelines recommend a series of variables to stratify patients into low, intermediate and high-risk categories (corresponding to an estimated one-year mortality rate of <5%, 5–10% and >10% respectively) which subsequently guides management [ 13 ]. These variables include an assessment of clinical function, World Health Organisation functional class (WHO-FC), exercise capacity and right ventricular function, with the WHO-FC segregating PAH patients into four categories based on their physical capacity impairment and dyspnea [ 13 , 14 ]. These variables are measured through a combination of imaging (echocardiography, cardiac magnetic resonance imaging), haemodynamics (e.g., right atrial pressure, cardiac index, mixed venous oxygen saturation), exercise testing (e.g., 6-minute walking test, cardiopulmonary exercise testing) and biochemical markers (markers of vascular dysfunction, myocardial stress, low cardiac output and secondary organ damage) [ 13 , 14 ]. Other factors such as age, sex, PAH subtype and symptomatic features of heart failure should be considered in prognostic evaluation, although their importance is largely determined on a case-by-case basis supplemented with clinical expertise [ 13 ].
Prognostic risk stratification of pulmonary arterial hypertension. Adapted from 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension [ 13 ].
RHF = right heart failure; WHO = World Health Organisation; BNP = brain natriuretic peptide; NT-proBNP = N-terminal pro-brain natriuretic peptide; CMR = cardiac magnetic resonance; VO 2 = oxygen consumption; pred = predicted; VE/VCO 2 = minute ventilation/carbon dioxide production; RA = right atrium; RAP = right atrial pressure; CI = cardiac index; SvO 2 = mixed venous oxygen saturation; a Occasional syncope—syncope that occurs only with high intensity exercise. b Recurrent syncope—syncope that occurs with moderate to low intensity exercise.
3. Genetics
Heritable PAH (hPAH), a subcategory of PAH, exhibits an autosomal dominant pattern of inheritance, with several associated germline gene mutations having been identified [ 15 ]. These mutations primarily occur in the genes encoding the transforming growth factor β (TGF-β) receptor superfamily, namely BMPRII [ 16 ], activin receptor-like kinase 1 ( ALK1 ) [ 17 ], mothers against decapentaplegic homolog 9 ( SMAD9 ) and endoglin 1 ( ENG ) [ 16 , 17 , 18 ]. Approximately 70% of hPAH patients [ 15 ], and a minority (10–40%) of patients with apparently sporadic iPAH [ 19 ], have mutations in the BMPRII ; in comparison to mutations in ALK 1 , SMAD9 and ENG , which collectively appear in only 5% of the hPAP population [ 15 , 19 , 20 ]. Though hPAH was previously considered to exhibit genetic anticipation (i.e., earlier age of onset and death in consecutive generations), recent data has challenged this conclusion [ 21 ].
Diagnosis of hPAH is complicated by the incomplete penetrance of BMPRII mutations, with only 20% of individuals possessing disease-associated variants developing the condition [ 21 ]. Furthermore, the variable expressivity and female predominance of these gene variants reveal the combination of genetic, genomic and environmental factors in PAH pathogenesis [ 21 , 22 ].
The most commonly studied gene mutation in relation to PAH pathogenesis is with BMPRII . Animal models demonstrate that reduced BMPRII activity in pulmonary vascular endothelial cells increases the incidence of apoptosis, leading to vascular remodelling and ultimately PAH [ 23 , 24 ]. Additionally, improving BMPRII expression in mice models through microRNA inhibition limits endothelial dysfunction and attenuates hypoxia-induced PAH [ 25 ].
Though genetic testing for hPAH is available, this service should be offered by trained individuals to those patients with iPAH considered to be sporadic or induced by anorexigens and to patients with a family history of PAH [ 13 ]. Ethical principles of genetic testing must include, among others, preserving patient and family autonomy, avoiding harm, and allowing equal access to genetic counselling for all patients. As outlined previously, the variable penetrance and expressivity of the BMPRII mutations may cause genetic testing to identify variants of unknown clinical significance, thereby causing unnecessary anxiety. Nonetheless, genetic testing is available which involves initial testing of only BMPRII variants, with negative results prompting further investigation of rarer pathogenic mutations (e.g., ALK1 and ENG ) [ 13 ].
4. Pathophysiology
PAH may be idiopathic or secondary to various conditions, but regardless of the underlying aetiology, patients exhibit similar pathological changes which include enhanced pulmonary arteriole contractility, endothelial dysfunction, remodelling and proliferation of both endothelial and smooth muscle cells, and in situ thrombi [ 5 ]. The physiological outcome of these disturbances is the partial occlusion of small pulmonary arteries, eventuating in increased PVR, subsequent right ventricular failure and death [ 5 ].
Underpinning these progressive pulmonary vascular defects is the disruption of three key signalling pathways outlined in Figure 1 : nitric oxide (NO), prostacyclin (PGI 2 ) and thromboxane A 2 (TXA 2 ), and endothelin-1 (ET-1) [ 26 ]. Broadly speaking, PAH is caused by impaired vasodilation from reduced PGI 2 production (cyclooxygenase-2 dysregulation) and NO synthase (eNOS) function, with concurrent vasoconstrictive and mitogenic effects of an upregulated ET-1 signalling system [ 26 , 27 ]. A mechanistic understanding of these three pathways has prompted rapid development in the quantity and efficacy of targeted pharmacological therapies for PAH.

The key abnormal pathways targeted in the pharmacological treatment of pulmonary arterial hypertension and the mechanism of action for contemporary drugs. The dashed line from ET B denotes action of endothelial ET B activation via NO and PGI 2 production. Adapted from Prior et al. MJA 2016 [ 28 ].
4.1. Nitric Oxide Pathway
Nitric oxide is produced in endothelial cells by eNOS, which, in the presence of oxygen, NADPH and other cofactors, catalyses the oxidation of l -arginine to l -citrulline. NO diffuses into the underlying pulmonary vascular smooth muscle cells (PVSMC) and binds to soluble guanylate cyclase (sGC), which in turn, converts guanosine triphosphate (GTP) to cyclic guanosine monophosphate (cGMP). The subsequent activation of downstream cGMP-dependent protein kinases (PKG) results in pulmonary vasodilation. Additionally, NO inhibits PVSMC proliferation, platelet aggregation and thrombosis, collectively maintaining normal healthy pulmonary vasculature.
In PAH, there is decreased bioavailability of NO, causing vasoconstriction and increased smooth muscle cell proliferation, inflammation and thrombosis. Although these pathological changes were initially attributed to observed reductions of eNOS expression amongst PAH patients, more recent studies have demonstrated similar outcomes from persistent eNOS activation in mice and human models [ 27 , 29 ]. A potential explanation for this apparent contradiction is the role of reactive oxygen species (ROS), particularly tetrahydrobiopterin (BH 4 ), in the enzymatic uncoupling of eNOS, thereby accounting for the pathogenesis of endothelial dysfunction, vasoconstriction and vascular remodelling in these models [ 30 ].
There are currently two approved drug classes acting on the nitric oxide pathway: phosphodiesterase 5 inhibitors (PDE-5i) and guanylate cyclase (GC) stimulators. PDE-5i prevent the degradation of cGMP, thereby increasing its plasma concentration and promoting the vasodilatory and antiproliferative effects of NO. GC stimulators act directly on sGC, even in the absence of NO, conferring similar increases in cGMP concentration.
4.2. Prostacyclin-Thromboxane A 2 Pathway
Prostacyclins are produced in endothelial cells from arachidonic acid via cyclooxygenase and prostacyclin synthase. PGI 2 binds to specific I-prostanoid (IP) receptors in smooth muscle cells, thereby activating adenylate cyclase. This enzyme converts adenosine triphosphate (ATP) to cyclic adenosine monophosphate (cAMP), which ultimately causes smooth muscle relaxation and subsequent vasodilation. Prostacyclin inhibits platelet aggregation, attenuates smooth muscle proliferation, and produces anti-inflammatory and antithrombotic effects.
In PAH, the pathway shifts towards an alternative product, thromboxane A 2 , leading to platelet aggregation, vasoconstriction and proliferation [ 31 ]. The aetiological role of prostacyclin is evidenced by animal models, with IP-knockout mice exhibiting severe PAH and subsequent vascular remodeling when subjected to chronic hypoxia [ 32 ]. Furthermore, patients with PAH have reduced production of prostacyclins as well as reduced expression of prostacyclin receptor and prostacyclin synthase [ 33 ].
The clinical drugs developed for PAH therapy that act on the prostacyclin pathway are the prostacyclin analogues and receptor agonists.
4.3. Endothelin-1 Pathway
Endothelin-1 (ET-1) is a peptide that acts as a potent vasoconstrictor [ 34 ]. ET-1 is produced on endothelial cell membranes from the precursor peptide big-endothelin-1 by endothelin-converting enzymes. ET-1 activates ET A and ET B : two G-protein coupled receptors. ET A is found on vascular smooth muscle cells, and promotes vasoconstriction, hypertrophy, proliferation, cell migration and fibrosis when activated. ET B is located on both vascular smooth muscle and endothelial cell surfaces. On smooth muscle, activation of ET B causes vasoconstriction whilst on endothelial surfaces, ET B activates NO and prostacyclin production, causing vasodilation and anti-proliferation [ 35 ].
During PAH, there is an increase in expression of ET A and smooth muscle ET B , but reduced expression of endothelial ET B [ 35 ]. In addition, PAH patients exhibit increased ET-1 concentrations in their plasma and pulmonary vascular endothelial cells [ 36 , 37 ]. The pathological alterations to this pathway are counteracted by endothelin receptor antagonists (ERAs), which are available as ET A selective or dual-action on ET A and ET B receptors.
5. Management of PAH
Treatment of PAH has progressed significantly over the past few decades in both its complexity and efficacy [ 38 , 39 ]. The aim of therapy is to achieve a low-risk status (maintaining WHO-FC II if possible) to preserve patient function, quality of life and minimise mortality risk [ 13 ]. This is generally achieved by optimising the patient’s six-minute walk distance (6MWD); however, current target thresholds rely upon cohort studies and expert consensus [ 40 , 41 ].
PAH management involves a stepwise pragmatic approach from general supportive treatment up to targeted pharmacological interventions as outlined in Figure 2 . Selected patients undergo vasoreactivity testing, with those that exhibit sufficient vasodilation subsequently being commenced on regular high-dose calcium channel blockers (CCB) [ 13 ]. Patients who either fail to qualify for vasoreactivity testing or demonstrate inadequate response are then commenced on targeted monotherapy or combination therapies that counteract the pathophysiology of the condition.

The European Society of Cardiology and the European Respiratory Society (ESC/ERS) evidence-based treatment algorithm for pulmonary arterial hypertension patients. Adapted from ECS/ERS guidelines for the diagnosis and treatment of pulmonary hypertension [ 13 ].
5.1. General and Supportive Therapies
Supportive care remains the mainstay of PAH management despite insufficient and heterogeneous data supporting these practices [ 13 ]. These measures (supervised physical exercise, pregnancy avoidance, appropriate birth control, oxygen supplementation, diuretics, digoxin, oral anticoagulation, psychosocial support, genetic counselling) serve to provide symptomatic relief or minimise the morbidity associated with this condition.
Patients with PAH are recommended to engage in physical activity as permitted by their symptomatic control, with supervised rehabilitation performed at specialist centres for deconditioned patients [ 13 ]. The benefit of physical activity is elucidated by a randomised controlled trial (RCT) of 30 patients with optimal medical control, with the exercise group demonstrating improved exercise capacity, 6MWD, quality of life, functional status and oxygen consumption levels compared to non-training controls [ 42 ]. Subsequent RCTs have similarly found exercise training to improve fatigue levels, 6MWD and patient quality of life [ 43 , 44 ]. These recommendations are hampered by the limited study sample sizes (the largest having 183 patients), unknown prognostic implications and the lack of direct comparison between different exercise regimes.
Pregnancy, due to its associated high mortality and morbidity in PAH, is not recommended [ 13 , 45 ]. Additionally, pregnancy reduces the range of drugs available for PAH treatment since ERAs are formally contraindicated given their teratogenicity [ 46 , 47 ]. Recent registry data suggest treatment advancements may have partially ameliorated this burden, particularly amongst CCB-managed patients, but there are insufficient extensive, long-term trials to amend current pregnancy recommendations [ 48 ].
Appropriate birth control measures are an area of inquiry with less consensus. Barrier methods and progesterone are recommended and often used in combination, while oestrogens are used cautiously due to the risk of venous thromboembolism, pulmonary vascular effects and reduced efficacy due to drug–drug interactions with the ERA bosentan [ 49 ]. Should pregnancy continue despite education concerning high maternal and fetal risks, patients should receive disease-specific therapy and elective delivery with support from a PAH specialty team [ 50 ].
Hypoxia causes significant pulmonary arterial vasoconstriction and oxygen therapy has been demonstrated to reduce PVR in PAH patients [ 51 ]. However, there are no long-term randomised trials that evaluate the effects of oxygen, but expert consensus recommends sufficient supplementation to maintain saturation levels above 90% [ 52 ].
Optimising intravascular fluid status through the appropriate administration of diuretics reduces right ventricular (RV) dilatation, hepatic congestion, ascites and oedema. Despite this theoretical rationale, no RCTs have investigated the role of diuretics in PAH, thus the type and dosage of diuretics is largely left to clinical discretion [ 13 ].
Digoxin acutely improves cardiac output in iPAH patients, but its long-term use remains controversial since there is no data concerning chronic effects or clinical benefit [ 53 ]. Angiotensin converting enzyme-inhibitors, angiotensin receptor blockers, beta-blockers and ivabradine are not routinely recommended in the absence of associated comorbidities [ 13 ].
Given observed abnormalities in coagulation and fibrinolytic pathways in PAH patients, oral anticoagulation is currently recommended in patients with iPAH, hPAH or PAH secondary to anorexigens, based primarily upon short-term, single-centre retrospective studies [ 13 , 54 , 55 , 56 , 57 , 58 ]. However, the limited registry and RCT evidence available is inconclusive, heterogeneous and to-date has not examined the role of the new oral anticoagulant agents [ 54 , 57 , 58 ]. Anticoagulation is not recommended in other subtypes of PAH [ 13 ].
Multidisciplinary teams at specialised PH clinics provide a holistic approach to manage all aspects of patient care. Psychosocial counselling and patient support groups are effective for minimising the psychological, social, financial, spiritual and emotional impact on patients and their families [ 59 , 60 ]. As previously discussed, the option to provide genetic counselling to eligible patients is offered when medically and ethically appropriate [ 13 ].
5.2. Calcium Channel Blockers
Calcium channel blockers (CCB) inhibit the inflow of calcium into smooth muscle cells, thereby causing vasodilation. Most PAH patients demonstrate minimal response to acute vasodilatory challenges during right heart catheterisation testing; however, some patients demonstrate vasodilation to normal or near-normal pressures. In some of these patients, ongoing high-dose CCBs can have dramatic improvements with sustained haemodynamics to near physiological values [ 61 ]. Long-term response to CCB is most commonly observed in iPAH patients [ 61 ]. In contrast, patients with PAH associated with CTD, HIV, Portopulmonary hypertension (porto-pulmonary-HTN) and Pulmonary veno-occlusive disease (PVOD) exhibit poor long-term CCB efficacy, even in the presence of positive vasoreactivity testing [ 62 ]. Therefore, the ESC/ERS guidelines recommend pulmonary vasoreactivity testing in iPAH, hPAH and drug induced PAH patients [ 13 ]. Patients with insufficient responses to high-dose CCBs should be commenced on additional PAH-specific therapies [ 13 ]. CCBs should not be administered in patients with negative vasodilatory results or those who have not undergone vasoreactivity studies, due to potential side effects of RV failure, hypotension and syncope [ 13 ].
5.3. Targeted Therapies
Specific therapies which address the underlying pathophysiology of PAH include phosphodiesterase-5 inhibitors (PDE-5i), guanylate cyclase (GC) stimulators, prostacyclin analogues, prostacyclin receptor agonists, and endothelin receptor antagonists (ERA). In comparison to historical treatment regimes, these targeted therapies have revolutionised modern PAH management [ 38 ], with several studies having demonstrated improved exercise capacity, WHO-FC and time to clinical worsening (TTCW) as summarised in Table 3 . The individual studies were not sufficiently powered to analyse mortality but a subsequent meta-analysis of RCTs demonstrated a 43% reduction in mortality rate [ 63 ].
Summary of randomised clinical trials of drugs approved for treatment of pulmonary arterial hypertension.
NS = not statistically significant; 6MWD = placebo corrected 6-minute walk distance; TTCW = time to clinical worsening; WHO-FC = World Health Organisation Functional Class; NYHA = New York Heart Association BNP = brain natriuretic peptide; PVR = pulmonary vascular resistance; TPR = total pulmonary resistance; CI = cardiac index; ERA = endothelin receptor antagonists; PDE-5i = phosphodiesterase type-5 inhibitors; PCA = prostacyclin receptor agonist; PAH = pulmonary arterial hypertension.
5.4. Phosphodiesterase-5 Inhibitors
Sildenafil is a selective PDE-5i which is predominantly prescribed orally but can be administered intravenously for long-term patients who are unable to tolerate oral formulations. The SUPER-1 (Sildenafil Use in Pulmonary Arterial Hypertension) trial randomly allocated 278 treatment-naïve PAH patients to either placebo or sildenafil for 12 weeks [ 82 ]. Sildenafil conferred significant improvements in 6MWD, WHO-FC and pulmonary artery pressures when compared to placebo, but there was no difference in clinical worsening incidence [ 82 ]. When extended to three years in the SUPER-2 trial, 60% and 46% of patients maintained or improved their WHO-FC and 6MWD respectively [ 84 ]. Addition of sildenafil to epoprostenol similarly improved 6MWD and TTCW [ 76 ]. Some adverse effects of sildenafil included headaches, flushing, epistaxis, dyspepsia and diarrhoea, ranging from mild to moderate in severity [ 82 ]. Since PDE-5i cause vasodilation, these drugs should be used cautiously in combination with other vasodilatory agents, particularly nitrates and CCBs.
Tadalafil, an alternative PDE-5i medication, possesses a superior pharmacokinetic profile in that it is dispensed once-daily in comparison to sildenafil which requires thrice-daily administration. In the PHIRST-1 (Pulmonary Arterial Hypertension and Response to Tadalafil) trial, 405 patients who were either treatment-naïve or on existing bosentan therapy, were randomised to receive placebo or tadalafil for 16 weeks [ 78 ]. The tadalafil arms of both the treatment-naïve and background therapy groups demonstrated significantly improved exercise capacity, symptomatic control, haemodynamics and reduced TTCW [ 78 ]. Tadalafil was well tolerated in the long-term, with sustained improvements in exercise capacity [ 85 ].
5.5. Guanylate Cyclase Stimulators
Riociguat directly activates sGC, thus promoting vasodilation. In PATENT-1 (Pulmonary Arterial Hypertension Soluble Guanylate Cyclase-Stimulator Trial), 443 patients who were either treatment-naïve or had existing PAH treatment were randomised to placebo or riociguat, with the latter group demonstrating favourable outcomes regarding 6MWD, haemodynamics, WHO-FC and TTCW [ 77 ]. On sub-analysis, improved exercise capacity was also observed in patients on existing background therapy [ 77 ]. The long-term efficacy and safety profile of riociguat are currently being evaluated in PATENT-2, with interim analysis showing acceptable drug tolerability and continued improvements in 6MWD and WHO-FC compared to the PATENT-1 baseline [ 86 ]. The adverse effects of GC stimulators are similar to PDE-5i, including hypotension and syncope [ 77 ]. Combination therapy with PDE-5i was found to have a non-positive benefit-risk ratio due to increased hypotension and other drug-related side-effects [ 87 ].
5.6. Prostacyclin Analogues
Epoprostenol is a synthetic prostacyclin that can only be administered intravenously due to its short half-life (3–5 min) and limited stability at room temperature. Randomised trials have examined the benefit of continuous epoprostenol infusions in patients with iPAH [ 68 , 79 ] and PAH secondary to the scleroderma spectrum of disease [ 67 ], with this regimen producing improved symptomatic control, exercise capacity and haemodynamics [ 67 , 68 , 79 ]. Crucially, one study found improved survival rates among patients with severe iPAH—making epoprostenol the only therapy to-date to demonstrate a reduced mortality rate within a single RCT [ 68 ]. According to the ESC/ERS guidelines, a subsequent meta-analysis of the aforementioned randomised trials found a risk reduction in mortality of about 70% [ 13 , 67 , 68 , 79 ].
Treprostinil is a prostanoid analogue with similar properties to epoprostenol, but due to its longer half-life, it can be administered through multiple routes. A RCT comparing subcutaneous treprostinil to placebo demonstrated exercise capacity, symptomatic control and haemodynamics improved significantly, however infusion site pain was a common adverse event causing 8% of the population to withdraw from the study [ 81 ]. A RCT intended to investigate intravenous (IV) treprostinil was closed prematurely due to safety considerations [ 88 ]. Amongst the enrolled population, in 44 (35%) of the planned 126 participants, there were significant improvements in exercise capacity, symptoms and functional class [ 88 ]. Observational studies have found short-term IV treprostinil to be tolerable and produce a similar benefit profile to epoprostenol [ 89 , 90 ]. The TRIUMPH (Treprostinil Sodium Inhalation Used in the Management of Pulmonary Arterial Hypertension) trial investigated the addition of inhaled treprostinil to background therapy of either bosentan or sildenafil [ 83 ]. There was a significant improvement in 6MWD and quality of life, but minimal effect on functional class or TTCW [ 83 ]. The FREEDOM-C (Multicenter, Double-101 Blind, Randomized, Placebo-Controlled Study of the Efficacy and Safety of Oral Treprostinil Sustained Release Tablets in Subjects With Pulmonary Arterial Hypertension) [ 73 ] and FREEDOM-C2 [ 74 ] trials studied oral formulations of treprostinil in PAH patients on background bosentan and/or sildenafil therapy. Both studies demonstrated non-significant improvements in 6MWD [ 73 , 74 ] however a subsequent RCT with treatment-naïve patients observed a significant improvement [ 91 ].
Iloprost is available in IV, oral or aerosol formulations, although the effects of oral iloprost have not yet been studied. Inhaled iloprost, as demonstrated in the AIR (Aerosolized Iloprost Randomized) study, conferred significant improvements in symptomatic control, haemodynamics, WHO-FC and quality of life, though it was commonly associated with flushing and jaw pain [ 64 ]. Data concerning IV iloprost in PAH is scarce, with an observational study demonstrating limited clinical benefits [ 92 ].
5.7. Prostacyclin Receptor Agonists
Selexipag is an oral prostacyclin IP receptor agonist which produces vasodilatory and anti-proliferative effects. This agent was studied in the 71-week GRIPHON (Prostacyclin Receptor Agonist in Pulmonary Arterial Hypertension) study, which involved 1156 PAH patients with pre-existing monotherapy or dual therapy [ 75 ]. Time to first morbidity or mortality event was significantly reduced by 40%, though mortality rates were not significantly affected [ 75 ]. Selexipag significantly improved 6MWD, but was associated with headache, diarrhoea, nausea and jaw pain [ 75 ].
5.8. Endothelin Receptor Antagonists (ERA)
Bosentan is a dual-acting ERA, binding to both the ET A and ET B receptors. The BREATHE-1 (Bosentan Randomized Trial of Endothelin Antagonist Therapy) study commenced 213 treatment-naïve PAH patients on a 12-week bosentan regimen which conferred improvements in 6MWD, functional class and time to first worsening [ 69 ]. These findings were similarly demonstrated in a subsequent 24-week study, with both studies showing increased hepatic enzymes as the main adverse event [ 69 , 72 ]. COMPASS-2 is a more recent, 38-month trial involving 334 patients on background sildenafil therapy who were randomised to either bosentan or placebo [ 71 ]. This study did not meet its primary endpoint (time to first morbidity or mortality) nor its secondary endpoints (functional class and PAH hospital-related admissions) except for a significant improvement in 6MWD [ 71 ].
Ambrisentan selectively binds to ET A receptors, with minimal binding to vasodilatory endothelial ET B receptors [ 66 ]. ARIES-1 (Ambrisentan in Pulmonary Arterial Hypertension, Randomized, Double-Blind, Placebo-Controlled, Multicentre Efficacy Study) and ARIES-2 involved 202 and 192 treatment-naïve PAH patients respectively [ 66 ]. Ambrisentan elicited improvements in 6MWD, functional class and TTCW, but produced adverse effects including peripheral oedema, headache and nasal congestion [ 66 ].
Macitentan is a novel drug targeting ET A and ET B receptors. SERAPHIN (Study with an Endothelin Receptor Antagonist in Pulmonary Arterial Hypertension to Improve Clinical Outcome) compared macitentan to placebo in 742 PAH patients (who were either treatment-naïve or on background therapy) over an average of 100 weeks [ 80 ]. Macitentan significantly reduced the composite endpoint (morbidity and mortality events, PAH-related hospitalisations, 6MWD and WHO-FC), although the study was insufficiently powered to show significant mortality reduction in sub-analysis [ 80 ]. These benefits were observed in both treatment-naïve and background-treated patients [ 80 ].
5.9. Combination Therapies
Combination therapies have been increasingly recognised in the treatment of PAH as they allow the targeting of multiple signalling pathways. A meta-analysis performed by Galiè and colleagues showed a reduction in TTCW and improvements in clinical outcomes when sequential combination therapy was added following suboptimal response to monotherapy [ 13 , 93 ]. Although the meta-analysis demonstrated a reduction in mortality, the analysis was not sufficiently powered to achieve statistical significance [ 93 ]. A recent meta-analysis similarly demonstrated benefits of combination therapy over monotherapy [ 39 ].
BREATHE-2, an extension of BREATHE-1, was the first trial to explore combination therapy and involved 33 patients on existing epoprostenol treatment who were either randomised to placebo or bosentan [ 70 ]. BREATHE-2 failed to demonstrate significant differences in haemodynamics or clinical improvement, however these conclusions are limited by the study’s small sample size [ 70 ]. This has been investigated in subsequent studies, such as COMPASS-2 [ 71 ] and PACES [ 76 ], and some results can be inferred from subanalyses of other studies with patients on background therapy from baseline. Based on the findings of these studies, the ESC/ERS pulmonary hypertension guidelines outline several possible combination therapies for PAH [ 13 ].
The AMBITION (Ambrisentan and Tadalafil in Patients with Pulmonary Arterial Hypertension) trial was a pivotal study in combination therapy which directly investigated the role of initiating dual therapy in treatment-naïve patients by randomising these participants to either combination therapy or monotherapy of either ambrisentan or tadalafil for 73 weeks [ 65 ]. The primary endpoint (time to clinical failure event) was reached with both dual and monotherapy, with the former group exhibiting an additional 50% relative reduction compared to the latter [ 65 ]. This was the first study to provide a direct outcome comparison between monotherapy and combination therapy in treatment-naïve patients, thereby creating a new paradigm for the initial management of high-risk PAH.
5.10. Clinical Approach to PAH Treatment
The ESC/ERS guidelines recommend a stepwise pragmatic approach to PAH management as outlined in Figure 2 [ 13 ]. Recent long-term trials have revealed that targeted therapies confer significant improvements in clinical outcomes, thereby changing the therapeutic approach to PAH [ 39 , 63 ]. Risk assessment and treatment options should be considered at the time of diagnosis, with the aim of treatment to maintain low risk profiles [ 13 ]. Risk assessment, as discussed earlier, stratifies patients into low, intermediate or high risk of clinical worsening or death, although other prognostic factors should also be considered [ 13 ]. Patients with low or intermediate risk can be initiated on monotherapy, with regular monitoring of treatment response [ 13 ]. Results from the SERAPHIN and GRIPHON trials suggest there is benefit in adding either long-term macitentan or selexipag to background therapies in those patients who require additional treatment [ 75 , 80 ]. Conversely, for high-risk patients, the AMBITION trial highlighted the benefits of immediate combination therapy rather than a graduated, stepwise approach [ 65 ]. Often, IV epoprostenol is prioritised in high-risk patients given that this agent has been demonstrated to improve 3-month mortality rate of patients with severe PAH [ 13 , 68 ]. Patients refractory to treatment may be considered for lung transplantation or balloon atrial septostomy, the discussion of which exceeds the scope of this review.
Since bosentan induces cytochrome P450 isoenzymes CYP3A4 and CYP2C9, and sildenafil is metabolised by these same isoenzymes, care should you taken to avoid drug-drug interactions. Furthermore, PAH-targeted medications should be used cautiously in patients taking co-existing antihypertensives to avoid systemic hypotension.
5.11. Future Research
The past few decades have seen dramatic changes in the understanding and management of PAH, leading to improved symptomatic control, exercise tolerance and progression free survival [ 38 , 39 , 63 ]. However, there have been variable definitions in study endpoints—particularly TTCW, complicating its interpretation in meta-analyses [ 39 ]. This endpoint includes a collection of outcomes, including symptomatic worsening, lack of improvement, PAH-related hospital admissions, transplantation and mortality [ 94 ]. However, PAH-related hospitalisation, mortality and transplantation often follows clinical deterioration and hence TTCW may underestimate the true mortality and morbidity [ 39 ]. Further research should have a consistent definition in study endpoints and broader analysis of total PAH-related events [ 39 , 94 ]. Nevertheless, clinical worsening and mortality was still prevalent in these studies despite adequate therapy [ 39 , 63 ] indicating the need for identification of novel therapeutic agents. Research is currently underway to investigate the role of oestrogen [ 95 ], tyrosine kinase inhibitors [ 96 ] and BMPRII gene activation [ 97 ], (amongst many others) in the management of PAH.
6. Conclusions
Substantial advancements in the understanding and management of PAH have been made in the recent decades. The current understanding of PAH aetiology includes the nitric oxide, prostacyclin-thromboxane and endothelin-1 pathways. This has led to five classes of drugs which target these the pathways: namely phosphodiesterase-5 inhibitors, soluble guanylate cyclase inhibitors, prostacyclin analogues, prostacyclin receptor agonists and endothelin receptor antagonists. Recent long-term trials have shown evidence of progression-free survival with initial, or early, addition of these drugs, either in isolation or in combination with other drug classes. In particular, emerging evidence has favoured the use of initial combination therapies in high risk patients. These medications are recommended with general and supportive measures; however, the evidence supporting these practices is heterogeneous and inconclusive. Ongoing study into alternative pathways involved in PAH pathogenesis represent opportunities for future targets in PAH management.
Author Contributions
All authors were involved in the drafting of the manuscript. All authors agreed with the final version of this manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
- Associations
- Working Groups
- Escardio.org
- ESC eLearning
ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension
Esc clinical practice guidelines.
Pulmonary hypertension (PH) is a pathophysiological disorder, which may involve multiple clinical conditions and may be associated with a variety of cardiovascular and respiratory diseases. The complexity of managing PH requires a multi-faceted, holistic, and multidisciplinary approach, with active involvement of the patients with PH in partnership with clinicians. In recent years, substantial progress has been made in detecting and managing PH, and new evidence is timely integrated in this fourth edition of the ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. These comprehensive clinical practice guidelines cover the whole spectrum of PH with an emphasis on diagnosing and treating pulmonary arterial hypertension (PAH) and chronic thrombo-embolic pulmonary hypertension (CTEPH).
Guidelines and related materials are for use by individuals for personal or educational purposes. No commercial use is allowed. Re-use permission must be correctly obtained from the publisher .
ESC Guidelines are protected by copyright. As per opt out option under Article 4 §3 of EU Directive 2019/790, the ESC reserves all rights to license uses of ESC Guidelines to train or develop generative artificial intelligence (AI) models, large language models or other deep learning or machine learning models. Any use of ESC Guidelines in software tools, generative AI models, computational models, or algorithms in which ESC Guideline content is included, cited, or transformed in any way requires a formal license agreement.
Endorsed by cardiac societies
Main resources.

https://doi.org/10.1093/eurheartj/ehac237
Declarations of interest

Relive the presentation as it was live.
More resources

Implementation of the 2022 ESC/ERS Guidelines on the Diagnosis and Treatment of Pulmonary Hypertension

Essential insights on the Guidelines in under four minutes.

Educational material.

Have the ESC Pocket Guidelines with you all the time.

What do the ESC 2022 Guidelines mean for you and your patients?

Insights from the Guidelines Task Force Chairs.
- ESC Board and Committees
- ESC Policies
- Statutes & Reports
- ESC Press Office
- Press Releases
- ESC Congress
- ESC Cardio Talk
- Our Offices
- Conference Facilities
- Jobs in Cardiology
- Terms & Conditions
- Update your cookie settings
Need help?
Help centre Contact us
© 2024 European Society of Cardiology. All rights reserved.
Pulmonary Hypertension
What is pulmonary hypertension.
Pulmonary hypertension happens when the pressure in the blood vessels leading from the heart to the lungs is too high.
With pulmonary hypertension, the blood vessels to the lungs develop an increased amount of muscle in the wall of the blood vessels. The heart pumps blood from the right ventricle to the lungs to get oxygen. Because the blood does not have to travel very far, the pressure in this side of the heart and in the artery taking blood from the right ventricle to the lungs is normally low—usually much lower than systolic or diastolic blood pressure.
When the pressure in this artery gets too high, the arteries in the lungs can narrow and then the blood does not flow as well as it should, resulting in less oxygen in the blood. 1

During pulmonary hypertension, the arteries in the lungs can narrow and then the blood does not flow as well as it should, resulting in less oxygen in the blood.

What causes pulmonary hypertension?
Some common underlying causes of pulmonary hypertension include high blood pressure in the lungs’ arteries due to some types of congenital heart disease, connective tissue disease, coronary artery disease, high blood pressure, liver disease (cirrhosis), blood clots to the lungs, and chronic lung diseases like emphysema. Genetics also play a role.
Pulmonary hypertension can happen in association with many other diseases, such as lung disease and heart disease. Heart failure is common in pulmonary hypertension.
What are the risk factors for pulmonary hypertension?
- Pulmonary hypertension happens at all ages, including children, and its incidence increases with age.
- Pulmonary hypertension is more common among women, non-Hispanic black people, and people age 75 or older.
What are the signs and symptoms of pulmonary hypertension?
The symptoms of pulmonary hypertension during the initial stage of the disease are common to many other medical conditions (e.g., difficulty breathing, fatigue). This often results in a delayed diagnosis until more severe symptoms arise, such as dizziness, chest pain, ankle swelling, or feeling the heart race or pound (palpitations). 2,3
How is pulmonary hypertension treated?
There is no cure for pulmonary hypertension. However, there are many different types of treatments, including
- Inhaled medicine
- Medicine given through the veins under the skin
- Medicine to reduce swelling in the feet (diuretics)
- Oxygen therapy
How can I prevent pulmonary hypertension?
While not all pulmonary hypertension can be prevented, you can take steps to prevent it by making healthy lifestyle changes and managing high blood pressure, coronary heart disease, chronic liver disease, and chronic lung disease from tobacco use.
More Information
- MedlinePlus
- National Heart, Lung, and Blood Institute
- American Heart Association
- Pulmonary Hypertension Association
- George MG, Schieb L, Ayala C, Talwalkar A, Levant S. Pulmonary hypertension surveillance: United States, 2001 to 2010. Chest . 2014;146(2):476–95.
- Brown LM, Chen H, Halpern S, Taichman D, McGoon MD, Farber HW, et al. Delay in recognition of pulmonary arterial hypertension: factors identified from the REAL Registry. Chest . 2011;140(1):19–26.
- Deaño RC, Glassner-Kolmin C, Rubenfire M, Frost A, Visovatti S, McLaughlin VV, et al. Referral of patients with pulmonary hypertension diagnoses to tertiary pulmonary hypertension centers. JAMA Intern Med . 2013;173(10):887–93.
Division for Heart Disease and Stroke Prevention
- High Blood Pressure
- Cholesterol
- Million Hearts ®
- WISEWOMAN Program
- Physical Activity
Exit Notification / Disclaimer Policy
- The Centers for Disease Control and Prevention (CDC) cannot attest to the accuracy of a non-federal website.
- Linking to a non-federal website does not constitute an endorsement by CDC or any of its employees of the sponsors or the information and products presented on the website.
- You will be subject to the destination website's privacy policy when you follow the link.
- CDC is not responsible for Section 508 compliance (accessibility) on other federal or private website.
Pulmonary Hypertension: ED Presentation, Evaluation, and Management
- Jan 11th, 2021
- Rachel Bridwell
- categories: practice updates
Author: Rachel Bridwell, MD (@rebridwell, EM Resident Physician, San Antonio, TX) // Reviewed by: Brit Long, MD (@long_brit, EM Attending Physician, San Antonio, TX); Skyler Lentz, MD (@skylerlentz, Assistant Professor of Surgery (EM) and Medicine (Critical Care), University of Vermont Medical Center); Alex Koyfman, MD (@EMHighAK, EM Attending Physician, UTSW / Parkland Memorial Hospital)
Clinical Case #1
A 27 year old male presents to the ED with 6 months of cough and dyspnea. He reports recent bouts of hemoptysis. He has not traveled or been exposed to any high risk areas for tuberculosis. He states he had heart surgery as a baby but cannot recall what it was. He additionally reports previous bouts of epistaxis but none recently.
On exam his vitals are HR 105, BP 104/81, RR 24, 90% on room air, T 98.1
HEENT: No conjunctival pallor
Pulm: Increased work of breathing though no wheezes, rhonchi, rales
CV: Tachycardic, palpable S2, positive left parasternal heave
Integumentary: no telangiectasias
Clinical Case #2
A 57 year old male with a PMHx of pulmonary hypertension and congestive heart failure presents with shortness of breath and syncope. He recently ran out of his high dose sildenafil. He denies chest pain though states his symptoms worsen with walking. Denies recent cough, fever, abdominal pain. He is a daily tobacco user.
On exam his vitals are HR 115, BP 101/72, RR 28, 86% on room air, T 98.1
CV: Tachycardic, S3, no murmurs
Integumentary: No clubbing or pallor
B ackground
Pulmonary hypertension (PH), characterized by increased pulmonary vascular resistance and pulmonary arterial pressure, is associated with significant morbidity and mortality. The disease is heterogenous, with varying demographics and underlying etiologies, affecting 15-60 million individuals worldwide. 1 PH is defined by a mean pulmonary arterial pressure of greater than 25 mm Hg by right heart catheterization or inferred by echocardiogram. 2 Although rare, with an estimated 5-15 cases per 1 million adults, recent studies identify PH-associated complaints as responsible for 64,000 ED visits over a 5 year period. 2 In the United States, the most common cause of PH is left sided heart failure. 3,4 In 1973, the World Health Organization (WHO) defined a classification system for PH to design and implement international health standards. This was revised in 2013 to redefine groups 1-5 (Table 1). 5,6

Pulmonary Pathophysiology
In the setting of PH, physiologic derangements represent changes in pulmonary and systemic circulation, altering the pressures in each system. The pulmonary circulation is normally a low-resistance, low-pressure system, composed of thin-walled vessels capable of accommodating vast alterations in preload. In patients with chronic PH, pulmonary vascular resistance gradually increases with vascular remodeling of the pulmonary arteries. This remodeling involves vascular smooth muscle and endothelial cell proliferation, inflammation, and fibrosis. If remodeling occurs over a prolonged period of time, the right ventricle (RV) is able compensate, however, in the setting of RV dilation, tricuspid regurgitation becomes common. Beyond a specific point of RV distension, RV output decreases secondary to increased pulmonary pressure and impedance to RV outflow. This derangement sets in motion a vicious cascade of interventricular dependence in which bulging of the RV into the left ventricle (LV) decreases LV filling, subsequently decreasing cardiac output (Fig. 1). 7–10 As cardiac output falls, end organ perfusion suffers, further exacerbating the pathophysiology of the inherent underlying disease. 9
Figure 1. Pathophysiology of PH.

Alterations in pulmonary system pressure and ventricular wall thickness additionally reduce coronary artery perfusion. In a normal physiologic setting, the coronary arteries of the RV are perfused in both systole and diastole due to low RV wall tension as compared to the LV. 10 In PH, as RV pressures increase, RV perfusion falls until pulmonary artery pressures exceed systemic pressure, generating RV ischemia. In this under-perfused state, RV contractility declines, worsening RV overload. 8,11,12 The emergency management of PH focuses on many of these pathways to combat these physiologic derangements to improve patient hemodynamic and respiratory status.
ED Evaluation
Patient Presentation
Accounting for approximately 5 to 15 cases per one million ED visits, PH and its associated symptoms are non-specific. 4 Dyspnea is the most common presenting complaint, though patients also report fatigue, weakness, chest pain, and syncope. 5,13 Symptomatology in undiagnosed PH is insidious; many individuals experience dyspnea and fatigue, worsening over weeks to months, eventually limiting daily activities. Angina results from ischemic subendocardial injury from ventilation-perfusion mismatch or compression of the left main coronary artery by the pulmonary trunk. 13 Syncope is also associated with a poor prognosis, while hoarseness may occur due to compression of the recurrent laryngeal nerve by an enlarged pulmonary artery. 5,13 In advanced disease, individuals frequently present with symptoms of right heart failure (RHF): ascites, peripheral edema, and hemoptysis. 13,14 Emergency clinicians should evaluate for underlying precipitants of RHF including sepsis, unplanned withdrawal of PH therapy, medication non-compliance, pregnancy, pneumonia, anemia, and arrhythmias. 41, 15
In individuals absent a previous diagnosis of PH, initial history should include PH risk factors including: congenital heart disease, left-sided heart disease, valvular disease, pulmonary disease, connective tissue disease, liver disease, blood dyscrasias, thyroid disorders, dialysis-dependent renal disease, malignancy, stimulant use, and family history of PH. 13,16 If available, a prior echocardiogram should be reviewed for signs of PH. In patients diagnosed with PH, questions regarding current therapy and medication compliance are essential to guide treatment and specialty consultation; therapeutic side effects of PH medications should be considered diagnoses of exclusion.
Physical Examination Findings
Physical examination is unreliable for determining the presence of PH in early disease stages. In advanced PH, signs of RHF are commonly present: elevated jugular venous pressure, hepatojugular reflex, ascites, hepatomegaly, and peripheral edema. 5,13,16 On auscultation, an increased pulmonic component of the second heart sound (P2), an RV gallop, or the murmur of tricuspid regurgitation may be present. In PH patients, arrhythmias including atrial fibrillation, atrial flutter, and atrioventricular node re-entry tachycardias are common. 40 In one systematic review, authors found that although a number of the aforementioned signs had high specificities (88% for an S4 on inspiration, 85% for a loud P2 on inspiration, 84% for an RV lift on inspiration), their sensitivities were low (12%, 29%, and 21%, respectively). 17 The physical examination finding with the highest positive likelihood ratio was a loud P2 on inspiration (LR+ 1.9, 95% Credible Interval 1.2-3.1). 17 Patients with decompensated PH frequently present with hypotension, displaying signs of systemic hypoperfusion including diaphoresis, cool extremities, peripheral cyanosis, and tachycardia. Patients with a patent foramen ovale who suffer from PH may exhibit symptoms of right-to-left shunting, manifesting as systemic hypoxemia and cyanosis not corrected by supplemental oxygen. 18,19
An electrocardiogram (ECG) should be obtained which in PH will reveal right axis deviation and right ventricular hypertrophy (Figure 2). 15,20 In addition to a tachyarrhythmia, right atrial enlargement, and ST segment depression and T wave inversion in the precordial leads may show right heart strain. 16
Figure 2. ECG with evidence of pulmonary hypertension.

Given the numerous etiologies of PH, laboratory studies should be guided by the patient presentation, history, and examination findings. In patients with chronic PH, venous blood gas frequently demonstrates hypoxemia and respiratory alkalosis. 13,16 Brain Natriuretic Protein (BNP) measurement may be useful in narrowing the differential diagnosis of a patient presenting with dyspnea. 20 BNP levels > 400 pg/ml suggest heart failure, but do not exclude other underlying conditions. 21 Normal plasma BNP levels increase with age and are higher in women than in men. 22 In the setting of renal failure, BNP should be interpreted with caution as reduced clearance may lead to chronic elevation. 20 Evaluation of myocardial perfusion and end-organ function is critical in patients presenting with signs and symptoms consistent with heart failure, prompting laboratory evaluation of troponin, renal function panel, liver function panel, and lactate, as an elevated troponin and liver function tests portend a poor prognosis. 21 The degree of heart failure is reflected by elevations in AST, ALT, and total bilirubin signify a decreased cardiac index and increased central venous pressure. 22,23
Imaging narrows the differential diagnosis among etiologies of PH and guides resuscitation, which should include a chest radiograph and bedside echocardiography (Figure 3). Radiographic findings of PH include dilated pulmonary arteries, peripheral pruning, and an enlarged right atria and RV, while pleural effusions may reflect severe disease. 16 While chest radiography demonstrates high sensitivity (96.9%) and specificity (99.1%) for detecting severe PH, it lacks sufficient sensitivity for detecting mild PH. 24
In the setting of PH, bedside echocardiography will review right-sided pressure overload: right atrial enlargement, RV dilation (RV: LV > 1:1; normal < 0.6), increased RV free wall thickness (> 5-7 mm as measured at end-diastole by M-mode or 2D echocardiography from the parasternal long axis or subxiphoid view), end-systolic flattening of the intraventricular septum, and interventricular interdependence visualized as a “D”-shaped left ventricle in diastole (Figure 3). 25 Ultrasound assessment of the inferior vena cava (IVC) may be misleading in PH patients, especially those with mitral regurgitation or aortic stenosis (Group 2 PH), revealing plethora that may not reflect intravascular volume status. 10,26
Figure 3. Ultrasound depicting RV dilatation.

Computed Tomography (CT) plays an essential role in identifying potential etiologies underlying PH (Figure 4). A CT demonstrating right atrial enlargement, RV dilation main pulmonary/ascending aorta diameter ratio ³ 1 is suggestive of PH, with a positive predictive value of 96% (Figure 4). 27 On CT, the pulmonary trunk should be no larger than 2.8 cm at the level of its bifurcation. Measurements > 2.8 cm suggest PH with a sensitivity of 69%–87% and a specificity of 89%–100%. 28 For individuals with CTEPH, CT angiography may reveal thrombi in the pulmonary vasculature and identify shunts contributing to the patient’s presentation. In this patient population, non-enhanced CT may reveal a mosaic pattern of variable attenuation in the lung parenchyma with evidence of irregular pulmonary perfusion due to chronic thromboemboli.
Figure 4. CT of the chest demonstrating evidence of PH with a main pulmonary/ascending aorta diameter ratio ³ 1 and a pulmonary trunk of >2.8 cm.
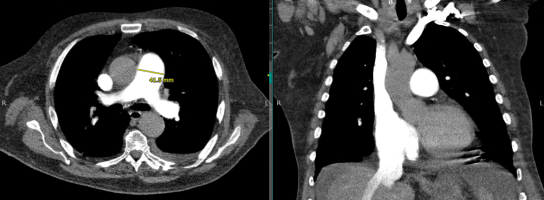
LV dysfunction is suggested on CT by the presence of a mosaic pulmonary perfusion pattern and pulmonary ground-glass opacities, demonstrating chronic pulmonary edema. The use of high-resolution CT in patients with PAH, without co-existing lung disease, should demonstrate normal lung parenchyma. Interstitial pulmonary abnormalities revealed by CT may point to an intrinsic lung disease as the etiology of the PH.
ED Management
The primary goal of the emergency clinician is the identification and treatment of the underlying etiologies of PH (e.g. alveolar hypoxia in COPD, hyperthyroidism, etc.). For individuals presenting with RHF, triggering factors should be addressed: antibiotic therapy administered for infections, transfusions given as indicated for anemia to maintain a hemoglobin of greater than 10 g/dL, and arrhythmias treated. 22,29 New-onset atrial fibrillation or flutter are common in PH patients. In the absence of randomized controlled trials, observational studies suggest improved mortality with a rhythm control strategy. 30 Rate control with calcium channel blockers or b-blockers is not advised, as these medications further impair RV function. 22 In particular, b-blockers should be avoided as they may result in systemic hypotension through negative inotropic and chronotropic effects. 31 Although digoxin may slow the ventricular rate in patients with supraventricular tachyarrhythmias associated with RV dysfunction, cardioversion is the preferred therapy for PH patients given this population’s propensity for digoxin toxicity. 32 Electrical cardioversion is favored as prolonged atrial arrhythmias in patients with PH are associated with rapid decompensation. Pharmacologic cardioversion with Class III agents, such as amiodarone and sotalol, have been reported. 33–35 While Class IC agents may theoretically be utilized, data in PH patients is lacking. In individuals with atrial arrhythmias lasting > 48 hours, anticoagulation is advised prior to cardioversion. 30
In PH patients who present due to an unexpected discontinuation in oral or IV PH therapy, every effort should be made to contact the patient’s PH specialist to initiate ED treatment and prevent acute decompensation. While prostacyclins, endothelin receptor agonists, and phosphodiesterase inhibitors may not be immediately available in the ED, initiating these therapies early in the ED course may help to stave off clinical instability, though optimizing oxygenation and circulation should take priority. 36–38 In individuals with PH, supplemental oxygen is indicated to maintain an oxygen saturation > 90%. 22,39 Hypercapnia should be avoided as it results in further pulmonary vasoconstriction. 6,41,22,39 Continuous non-invasive positive pressure ventilation may be considered, though fluid balance must be optimized prior to initiation to eliminate dangerous decreases in cardiac output. 22 Although data are limited in the setting of acute exacerbations, non-invasive positive pressure ventilation in Group 3 PH patients with no LV dysfunction is typically well tolerated and improves hypercapnia. 40,41 There are no current recommendations regarding the use of bilevel positive pressure ventilation in PH patients given the paucity of data. 610,41 High-flow nasal cannula (HFNC) is an alternative therapy that may improve hypoxemia, especially if patients are unable to tolerate the mask utilized for NIPPV.
Intubation should be avoided if possible, as the effect of sedatives and positive intrathoracic pressure may reduce preload, cardiac function and cause peripheral vasodilation, resulting in hypotension and cardiovascular collapse. 10 If intubation is required, etomidate is recommended for induction, given its limited effects on cardiac contractility and vascular tone. Hemodynamic optimization prior to intubation is recommended. If the patient is hemodynamically unstable, vasopressors should be initiated before attempts to establish a definitive airway are made. 21,42 Awake intubation with topical anesthetics is an alternative that should be considered given the reduced risk of hemodynamic decompensation as compared to rapid sequence intubation. Medications administered during rapid sequence induction will likely result in profound hemodynamic collapse, hypercarbia, hypoxemia, and acidosis. 10 Ventilator settings should target 6-8 mL/kg of ideal body weight and plateau pressures less than 30 cm H 2 O. Low positive end expiratory pressures should be utilized to minimize decreases in preload and increases in RV afterload. 8,21 Hypoxemia and hypercapnia should be avoided. 10,16,51,58
In the majority of cases, RV failure will be associated with fluid overload. 32 IV diuretics should be used cautiously to obtain a negative fluid balance, optimizing circulating blood volume and reducing RV preload, and thus improving cardiac output. 43 For patients not previously receiving oral diuretics, an initial dose of 20 – 40 mg IV furosemide is recommended for hypervolemia. 43 In individuals utilizing home diuretic therapy, the initial IV dose should be at least equivalent to the oral dose. 42 Consultation may be required for ultrafiltration in patients who are resistant to diuretic therapy. 42 In hypovolemic patients, volume should be delivered conservatively, with boluses of 250 mL over 15-30 minutes. 43
In the setting of hemodynamic instability, vasopressors should be initiated. Vasopressors increase aortic root pressure, increasing RV perfusion. 10 Norepinephrine is an effective first-line vasopressor for patients with PH. 43–45 Although norepinephrine primarily targets a 1 receptors, with limited b 1 stimulation to increase cardiac contractility, studies in heart failure patients have demonstrated improved RV myocardial oxygen delivery following administration (dose: 0.01-0.03mg/kg/min IV). 44 The addition of low dose vasopressin (0.01-0.03 U/min) may be considered if the aforementioned therapies fail to result in hemodynamic improvement. Low doses of vasopressin may cause a decrease in pulmonary vascular resistance while beneficially raising the systemic blood pressure, proving uniquely beneficial in this action. 46 Higher doses should be avoided (>0.08 U/min), which result in in pulmonary and coronary vasoconstriction. 44 ,75
Epinephrine may benefit PH patients given the combined alpha and beta stimulation which provides system vascular support with inotropy, though this may increase myocardial oxygen demand. To date, there have been no trials regarding epinephrine use in adult PH patients. In a pilot study of hemodynamically stable children, epinephrine demonstrated an increase in systemic vascular resistance, though it worsened the systolic pulmonary artery pressure: aortic pressure ratio. 47 Additionally, two patients experienced brief dysrhythmias, atrial bigeminy and ventricular bigeminy following administration of epinephrine. 47 Animal models suggests that epinephrine improves cardiac index through inotropy, with systemic vascular resistance increasing in a dose dependent fashion. 47,48 In contrast, sole a stimulation from phenylephrine should not be used in the unstable PH patient as it increases pulmonary vascular resistance, and may induce reflex bradycardia. 49 Dopamine should be also avoided given b 2 -mediated decreases in systemic vascular resistance and possible arrhythmias. 44
Inotropes increase the risk of tachyarrhythmias and should only be utilized in the setting of inadequate oxygen delivery, despite correction of abnormalities in RV preload and conditions causing RV ischemia. 44 If inotrope support is required, dobutamine is the agent of choice. 42,43 At low doses (5-10 mg/kg/min), dobutamine improves RV contractility and increases cardiac output through b 1 receptor stimulation. 42,43 Higher doses should be avoided, given increased stimulation of b 2 receptors, causing vasodilation and hypotension. 43 Milrinone, a selective PDE-3 inhibitor, is recommended for PH resulting from biventricular failure (0.375-0.75 mg/kg/min IV). Use in the ED may be limited given the requirement for pharmacy preparation and the drug’s slow onset of action. 43 Milrinone improves inotropy and pulmonary vasodilation, but similar to dobutamine, it may cause hypotension. 42,43
There are several other therapeutic options for patients with PH. Although sildenafil reduces RV afterload through pulmonary and systemic vasodilation, its use in critically ill patients in the ED is limited given the risk of hypotension. Patients who fail to respond to inotrope and vasopressor therapy should be considered for venoarterial extracorporeal membrane oxygenation. 43,49 Patients with PH have a high risk of sudden cardiac death and poor outcomes. Cardiopulmonary resuscitation outcomes in PH patients with RV failure are poor: in a cohort of 3,130 PH patients who required CPR, only 6% survived for more than 90 days. 21,50–52
Special Population – Prostacyclin Agonist Pump
A life-threatening emergency can occur if the patient has been prescribed IV epoprostenol or treprostinil and the IV catheter is removed or damaged, or the pump stops working. 53,54 A PH specialist should be consulted emergently, and if possible, the pump should not be turned off as this may result in sudden death. If there is a problem with the line, pump, or cassette, peripheral access should be obtained and the pump tubing connected directly to this access. 37,53,54 The line should not be primed or flushed, as a bolus of prostacyclin agonist may be delivered to the patient, resulting in fatal hypotension. The patient’s catheter should be inspected for drainage or surrounding cellulitis, which suggest infection. If administering medications or drawing laboratory studies, a second peripheral IV is required. An alternative infusion pump should not be utilized unless advised by the PH specialist. 53,54
Inhaled Therapies
With low systemic absorption, inhaled vasodilators, e.g. nitric oxide and epoprostenol, decrease pulmonary vascular resistance, bettering cardiac output. 55 These can be administered via endotracheal tube, NIPPV or HFNC, though depend on local hospital policy whether a closed circuit is required. 56 However, these should be avoided in patients with LV failure and only initiated in conjunction with PH experts. 55
Pulmonary Hypertension Crisis
Patients with PH may present unstable from an acute illness (e.g. sepsis) or become unstable after an intervention (e.g. intubation). The emergency clinician must have an approach to managing the patient with PH who becomes unstable to prevent the deadly cycle of hypotension, worsening pulmonary vascular resistance, RV ischemia worsening cardiac output leading to cardiovascular collapse. Below is a reference table to outline critical actions to take in a pulmonary hypertension crisis.

Disposition
If new onset PH, worsening or decompensated PH, or pump malfunction with prostacyclin agonist contribute to the ED presentation, admission and consultation with the specialty team are warranted. A formal transthoracic echocardiography is recommended to characterize RV size and function, and measure pulmonary artery systolic pressure and tricuspid jet. 14,16
Key Points:
-Myriad of chief complaints can be pulmonary hypertension; try to determine what is triggering the acute decompensation .
– Elevations in troponin and liver function tests portend a poor prognosis.
– Point of care ultrasound may be useful in guiding acute resuscitation, though evaluation of IVC may not reflect intravascular volume.
– Avoid hypoxemia and hypercarbia and maintain right ventricular preload support .
-Most patients will be admitted .
– Avoid intubating these patients if at all possible.
– Restart PAH meds if discontinued .
References / Further Reading
- Delcroix M, Howard L. Pulmonary arterial hypertension: The burden of disease and impact on quality of life. Eur Respir Rev . 2015;24(138):621-629. doi:10.1183/16000617.0063-2015
- Galiè N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J . 2016;37(1):67-119. doi:10.1093/eurheartj/ehv317
- Stein PD, Matta F, Hughes PG. Scope of Problem of Pulmonary Arterial Hypertension. Am J Med . 2015;128(8):844-851. doi:10.1016/j.amjmed.2015.03.007
- Rich JD, Rich S. Clinical diagnosis of pulmonary hypertension. Circulation . 2014;130(20):1820-1830. doi:10.1161/CIRCULATIONAHA.114.006971
- Rose-Jones L, Mclaughlin V. Pulmonary Hypertension: Types and Treatments. Curr Cardiol Rev . 2014;11(1):73-79. doi:10.2174/1573403×09666131117164122
- Wilkins MR, Wharton J, Grimminger F, Ghofrani HA. Phosphodiesterase inhibitors for the treatment of pulmonary hypertension. Eur Respir J . 2008;32(1):198-209. doi:10.1183/09031936.00124007
- Santamore WP, Dell’Italia LJ. Ventricular interdependence: Significant left ventricular contributions to right ventricular systolic function. Prog Cardiovasc Dis . 1998;40(4):289-308. doi:10.1016/S0033-0620(98)80049-2
- Green EM, Givertz MM. Management of acute right ventricular failure in the intensive care unit. Curr Heart Fail Rep . 2012;9(3):228-235. doi:10.1007/s11897-012-0104-x
- Maeder MT, Holst DP, Kaye DM. Tricuspid Regurgitation Contributes to Renal Dysfunction in Patients With Heart Failure. J Card Fail . 2008;14(10):824-830. doi:10.1016/j.cardfail.2008.07.236
- Wilcox SR, Kabrhel C, Channick RN. Pulmonary hypertension and right ventricular failure in emergency medicine. Ann Emerg Med . 2015;66(6):619-628. doi:10.1016/j.annemergmed.2015.07.525
- Watts JA, Marchick MR, Kline JA. Right Ventricular Heart Failure From Pulmonary Embolism: Key Distinctions From Chronic Pulmonary Hypertension. J Card Fail . 2010;16(3):250-259. doi:10.1016/j.cardfail.2009.11.008
- Zamanian RT, Haddad F, Doyle RL, Weinacker AB. Management strategies for patients with pulmonary hypertension in the intensive care unit. Crit Care Med . 2007;35(9):2037-2050. doi:10.1097/01.CCM.0000280433.74246.9E
- Sztrymf B, Souza R, Bertoletti L, et al. Prognostic factors of acute heart failure in patients with pulmonary arterial hypertension. Eur Respir J . 2010;35(6):1286-1293. doi:10.1183/09031936.00070209
- Medarov BI, Judson MA. The role of calcium channel blockers for the treatment of pulmonary arterial hypertension: How much do we actually know and how could they be positioned today? Respir Med . 2015;109(5):557-564. doi:10.1016/j.rmed.2015.01.004
- Maisel A. B-type natriuretic peptide levels: Diagnostic and prognostic in congestive heart failure: What’s next? Circulation . 2002;105(20):2328-2331. doi:10.1161/01.CIR.0000019121.91548.C2
- Doust J, Lehman R, Glasziou P. The role of BNP testing in heart failure. Am Fam Physician . 2006;74(11):1893-1898. http://www.ncbi.nlm.nih.gov/pubmed/17168346. Accessed November 21, 2019.
- Colman R, Whittingham H, Tomlinson G, Granton J. Utility of the Physical Examination in Detecting Pulmonary Hypertension. A Mixed Methods Study. Bogaard H, ed. PLoS One . 2014;9(10):e108499. doi:10.1371/journal.pone.0108499
- Homma S, Di Tullio MR. Patent foramen ovale and stroke. J Cardiol . 2010;56(2):134-141. doi:10.1016/j.jjcc.2010.05.008
- Vaidya K, Khandkar C, Celermajer D. Current management aspects in adult congenital heart disease: Non-surgical closure of patent foramen ovale. Cardiovasc Diagn Ther . 2018;8(6):739-753. doi:10.21037/cdt.2018.09.09
- Redfield MM, Rodeheffer RJ, Jacobsen SJ, Mahoney DW, Bailey KR, Burnett JC. Plasma brain natriuretic peptide concentration: Impact of age and gender. J Am Coll Cardiol . 2002;40(5):976-982. doi:10.1016/S0735-1097(02)02059-4
- Hoeper MM, Granton J. Intensive care unit management of patients with severe pulmonary hypertension and right heart failure. Am J Respir Crit Care Med . 2011;184(10):1114-1124. doi:10.1164/rccm.201104-0662CI
- Barst RJ, McGoon M, Torbicki A, et al. Diagnosis and differential assessment of pulmonary arterial hypertension. In: Journal of the American College of Cardiology . Vol 43. Elsevier USA; 2004:S40-S47. doi:10.1016/j.jacc.2004.02.032
- van Deursen VM, Damman K, Hillege HL, van Beek AP, van Veldhuisen DJ, Voors AA. Abnormal Liver Function in Relation to Hemodynamic Profile in Heart Failure Patients. J Card Fail . 2010;16(1):84-90. doi:10.1016/j.cardfail.2009.08.002
- Miniati M, Monti S, Airò E, et al. Accuracy of chest radiography in predicting pulmonary hypertension: A case-control study. Thromb Res . 2014;133(3):345-351. doi:10.1016/j.thromres.2013.12.019
- Markley RR, Ali A, Potfay J, Paulsen W, Jovin IS. Echocardiographic Evaluation of the Right Heart. J Cardiovasc Ultrasound . 2016;24(3):183. doi:10.4250/jcu.2016.24.3.183
- Feissel M, Michard F, Faller JP, Teboul JL. The respiratory variation in inferior vena cava diameter as a guide to fluid therapy. Intensive Care Med . 2004;30(9):1834-1837. doi:10.1007/s00134-004-2233-5
- Shen Y, Wan C, Tian P, et al. CT-base pulmonary artery measurementin the detection of pulmonary hypertension. Med (United States) . 2014;93(27). doi:10.1097/MD.0000000000000256
- Kuriyama K, Gamsu G, Stern RG, Cann CE, Herfkens RJ, Brundage BH. CT-determined pulmonary artery diameters in predicting pulmonary hypertension. Invest Radiol . 1984;19(1):16-22. doi:10.1097/00004424-198401000-00005
- Ruiter G, Lankhorst S, Boonstra A, et al. Iron deficiency is common in idiopathic pulmonary arterial hypertension. Eur Respir J . 2011;37(6):1386-1391. doi:10.1183/09031936.00100510
- Wanamaker B, Cascino T, McLaughlin V, Oral H, Latchamsetty R, Siontis KC. Atrial arrhythmias in pulmonary hypertension: Pathogenesis, prognosis and management. Arrhythmia Electrophysiol Rev . 2018;7(1):43-48. doi:10.15420/aer.2018.3.2
- Perros F, de Man FS, Bogaard HJ, et al. Use of β-Blockers in Pulmonary Hypertension. Circ Hear Fail . 2017;10(4):e003703. doi:10.1161/CIRCHEARTFAILURE.116.003703
- Patel R, Aronow WS, Patel L, et al. Treatment of pulmonary hypertension. Med Sci Monit . 2012;18(4):RA31-RA39. doi:10.12659/MSM.882607
- Cannillo M, Grosso Marra W, Gili S, et al. Supraventricular Arrhythmias in Patients with Pulmonary Arterial Hypertension. Am J Cardiol . 2015;116(12):1883-1889. doi:10.1016/j.amjcard.2015.09.039
- Olsson KM, Nickel NP, Tongers J, Hoeper MM. Atrial flutter and fibrillation in patients with pulmonary hypertension. Int J Cardiol . 2013;167(5):2300-2305. doi:10.1016/j.ijcard.2012.06.024
- Wen L, Sun ML, An P, et al. Frequency of supraventricular arrhythmias in patients with idiopathic pulmonary arterial hypertension. Am J Cardiol . 2014;114(9):1420-1425. doi:10.1016/j.amjcard.2014.07.079
- Duarte JD, Hanson RL, Machado RF. Pharmacologic treatments for pulmonary hypertension: Exploring pharmacogenomics. Future Cardiol . 2013;9(3):335-349. doi:10.2217/fca.13.6
- Hohsfield R, Archer-Chicko C, Housten T, Harris Nolley S. Pulmonary arterial hypertension emergency complications and evaluation: Practical guide for the advanced practice registered nurses in the emergency department. Adv Emerg Nurs J . 2018;40(4):246-259. doi:10.1097/TME.0000000000000210
- Palevsky HI, Fishman AP. Chronic cor pulmonale. Etiology and management. JAMA . 1990;263(17):2347-2353. http://www.ncbi.nlm.nih.gov/pubmed/2182919. Accessed November 20, 2019.
- Galiè N, Rubin L, Hoeper M, et al. Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): a double-blind, randomised controlled trial. Lancet . 2008;371(9630):2093-2100. doi:10.1016/S0140-6736(08)60919-8
- Parola D, Romani S, Petroianni A, Locorriere L, Terzano C. Treatment of acute exacerbations with non-invasive ventilation in chronic hypercapnic COPD patients with pulmonary hypertension. Eur Rev Med Pharmacol Sci . 2012;16(2):183-191. http://www.ncbi.nlm.nih.gov/pubmed/22428469. Accessed November 23, 2019.
- Bento AM, Cardoso LF, Tarasoutchi F, Sampaio RO, Kajita LJ, Lemos Neto PA. Hemodynamic effects of noninvasive ventilation in patients with venocapillary pulmonary hypertension. Arq Bras Cardiol . 2014;103(5):410-417. doi:10.5935/abc.20140147
- Pritts CD, Pearl RG. Anesthesia for patients with pulmonary hypertension. Curr Opin Anaesthesiol . 2010;23(3):411-416. doi:10.1097/ACO.0b013e32833953fb
- Savale L, Weatherald J, Jaïs X, et al. Acute decompensated pulmonary hypertension. Eur Respir Rev . 2017;26(146). doi:10.1183/16000617.0092-2017
- Harjola VP, Mebazaa A, Čelutkiene J, et al. Contemporary management of acute right ventricular failure: A statement from the Heart Failure Association and the Working Group on Pulmonary Circulation and Right Ventricular Function of the European Society of Cardiology. Eur J Heart Fail . 2016;18(3):226-241. doi:10.1002/ejhf.478
- Bertolissi M, Bassi F, Da Broi U. Norepinephrine can be useful for the treatment of right ventricular failure combined with acute pulmonary hypertension and systemic hypotension. A case report. Minerva Anestesiol . 2001;67(1-2):79-84.
- Price LC, Wort SJ, Finney SJ, Marino PS, Brett SJ. Pulmonary vascular and right ventricular dysfunction in adult critical care: Current and emerging options for management: A systematic literature review. Crit Care . 2010;14(5):1-22. doi:10.1186/cc9264
- Siehr SL, Feinstein JA, Yang W, Peng LF, Ogawa MT, Ramamoorthy C. Hemodynamic effects of phenylephrine, vasopressin, and epinephrine in children with pulmonary hypertension: A pilot study. Pediatr Crit Care Med . 2016;17(5):428-437. doi:10.1097/PCC.0000000000000716
- Hyldebrandt JA, Sivén E, Agger P, et al. Effects of milrinone and epinephrine or dopamine on biventricular function and hemodynamics in an animal model with right ventricular failure after pulmonary artery banding. Am J Physiol Circ Physiol . 2015;309(1):H206-H212. doi:10.1152/ajpheart.00921.2014
- Ventetuolo CE, Klinger JR. Management of acute right ventricular failure in the intensive care unit. Ann Am Thorac Soc . 2014;11(5):811-822. doi:10.1513/AnnalsATS.201312-446FR
- Demerouti EA, Manginas AN, Athanassopoulos GD, Karatasakis GT. Complications leading to sudden cardiac death in pulmonary arterial hypertension. Respir Care . 2013;58(7):1246-1254. doi:10.4187/respcare.02252
- Delcroix M, Naeije R. Optimising the management of pulmonary arterial hypertension patients: Emergency treatments. Eur Respir Rev . 2010;19(117):204-211. doi:10.1183/09059180.00004910
- Hoeper MM, Galié N, Murali S, et al. Outcome after cardiopulmonary resuscitation in patients with pulmonary arterial hypertension. Am J Respir Crit Care Med . 2002;165(3):341-344. doi:10.1164/ajrccm.165.3.200109-0130c
- Ruan CH, Dixon RAF, Willerson JT, Ruan KH. Prostacyclin therapy for pulmonary arterial hypertension. Texas Hear Inst J . 2010;37(4):391-399. doi:10.1007/978-1-60327-075-5_12
- (No Title). https://phassociation.org/wp-content/uploads/2017/02/school-resource-guide-Emergency-101-for-EMTs.pdf. Accessed November 21, 2019.
- Hill NS, Preston IR, Roberts KE. Inhaled therapies for pulmonary hypertension. Respir Care . 2015;60(6):794-802. doi:10.4187/respcare.03927
- Tremblay JA, Couture ÉJ, Albert M, et al. Noninvasive Administration of Inhaled Nitric Oxide and its Hemodynamic Effects in Patients With Acute Right Ventricular Dysfunction. J Cardiothorac Vasc Anesth . 2019;33(3):642-647. doi:10.1053/j.jvca.2018.08.004
Share this:

Leave a Reply Cancel reply
Your email address will not be published. Required fields are marked *
Save my name, email, and website in this browser for the next time I comment.
Notify me of follow-up comments by email.
Notify me of new posts by email.
WRITE FOR emDocs
We are actively recruiting both new topics and authors. This project is rolling and you can submit an idea or write-up at any time! Contact us at [email protected]
Algorithm of the month
FROM THE ARCHIVES
- emDOCs Podcast – Episode 100: Acute Chest Syndrome Part 1
EM@3AM: Brainstem Strokes
- Medical Malpractice Insights: Brain Abscess
Pediatric Oncologic Emergencies
Journal feed weekly wrap-up, emdocs in your mailbox.
Enter your email address to receive notifications of new posts by email.
Featured Articles
Emdocs podcast – episode 100:..., medical malpractice insights: brain..., emdocs podcast – episode 99: ..., em@3am: malignant/necrotizing otiti..., pocus findings of hemodynamically u..., medical malpractice insights: a tal....
emDocs is licensed under a Creative Commons Attribution 4.0 International License. Powered by Gomalthemes.
The effects of oxygenation on acute vasodilator challenge in pulmonary arterial hypertension
Affiliations.
- 1 Department of Medicine University of Colorado Anschutz Medical Campus Aurora Colorado USA.
- 2 Department of Biostatistics & Informatics, Colorado School of Public Health University of Colorado Anschutz Medical Campus Aurora Colorado USA.
- 3 Department of Medicine, Division of Pulmonary Sciences and Critical Care Medicine, Pulmonary Vascular Center University of Colorado Anschutz Medical Campus Aurora Colorado USA.
- 4 Department of Medicine, Division of Cardiology University of Colorado Anschutz Medical Campus Aurora Colorado USA.
- PMID: 38736893
- PMCID: PMC11088807
- DOI: 10.1002/pul2.12375
Identification of long-term calcium channel blocker (CCB) responders with acute vasodilator challenge is critical in the evaluation of patients with pulmonary arterial hypertension. Currently there is no standardized approach for use of supplemental oxygen during acute vasodilator challenge. In this retrospective analysis of patients identified as acute vasoresponders, treated with CCBs, all patients had hemodynamic measurements in three steps: (1) at baseline; (2) with 100% fractional inspired oxygen; and (3) with 100% fractional inspired oxygen plus inhaled nitric oxide (iNO). Those meeting the definition of acute vasoresponsiveness only after first normalizing for the effects of oxygen in step 2 were labeled "iNO Responders." Those who met the definition of acute vasoresponsiveness from a combination of the effects of 100% FiO 2 and iNO were labeled "oxygen responders." Survival, hospitalization for decompensated right heart failure, duration of CCB monotherapy, and functional data were collected. iNO responders, when compared to oxygen responders, had superior survival (100% vs. 50.1% 5-year survival, respectively), fewer hospitalizations for acute decompensated right heart failure (0% vs. 30.4% at 1 year, respectively), longer duration of CCB monotherapy (80% vs. 52% at 1 year, respectively), and superior 6-min walk distance. Current guidelines for acute vasodilator testing do not standardize oxygen coadministration with iNO. This study demonstrates that adjusting for the effects of supplemental oxygen before assessing for acute vasoresponsiveness identifies a cohort with superior functional status, tolerance of CCB monotherapy, and survival while on long-term CCB therapy.
Keywords: oxygen; pulmonary arterial hypertension; vasoreactivity.
© 2024 The Authors. Pulmonary Circulation published by John Wiley & Sons Ltd on behalf of Pulmonary Vascular Research Institute.

THOMAS A. PETERSON, MD, SEAN P. TURNER, MD, AND KATELYN A. DOLEZAL, MD
Am Fam Physician. 2024;109(5):441-446
Author disclosure: No relevant financial relationships.
Acute pericarditis is defined as inflammation of the pericardium and occurs in approximately 4.4% of patients who present to the emergency department for nonischemic chest pain, with a higher prevalence in men. Although there are numerous etiologies of pericarditis, most episodes are idiopathic and the cause is presumed to be viral. Diagnosis of pericarditis requires at least two of the following criteria: new or worsening pericardial effusion, characteristic pleuritic chest pain, pericardial friction rub, or electrocardiographic changes, including new, widespread ST elevations or PR depressions. Pericardial friction rubs are highly specific but transient, and they have been reported in 18% to 84% of patients with acute pericarditis. Classic electrocardiographic findings include PR-segment depressions; diffuse, concave, upward ST-segment elevations without reciprocal changes; and T-wave inversions. Transthoracic echocardiography should be performed in all patients with acute pericarditis to characterize the size of effusions and evaluate for complications. Nonsteroidal anti-inflammatory drugs are the first-line treatment option. Glucocorticoids should be reserved for patients with contraindications to first-line therapy and those who are pregnant beyond 20 weeks' gestation or have other systemic inflammatory conditions. Colchicine should be used in combination with first- or second-line treatments to reduce the risk of recurrence. Patients with a higher risk of complications should be admitted to the hospital for further workup and treatment.
Acute pericarditis, or inflammation of the pericardium, has numerous etiologies and often produces a characteristic pleuritic chest pain. This article reviews patient-oriented evidence to guide the diagnosis and management of acute pericarditis.
Epidemiology
In one small study, acute pericarditis was diagnosed in 4.4% of patients admitted to the emergency department with nonischemic chest pain; it accounts for 0.2% of cardiovascular hospital admissions. 1 , 2 The exact incidence of acute pericarditis is difficult to estimate because epidemiologic studies are lacking, and mild cases likely resolve without being formally diagnosed. 3 , 4
Acute pericarditis occurs mostly in adult patients, with a mean age in the 50s. 3 – 9 Hospital registry data suggest that men are more likely to be affected by acute pericarditis than women, with incidence ratios of 1.7 to 2.0 in men to 1.0 in women. 2 – 5
Acute pericarditis is typically a result of systemic disease or related to processes involving the pericardium 3 , 6 , 9 – 15 ( eTable A ) .
Despite advances in diagnostic testing, more than 50% of episodes are idiopathic and the etiology is presumed to be viral. 3 , 5 Pericarditis after cardiac injury is emerging as the second leading cause of pericarditis and occurs in 9% to 33% of patients. 3 , 5 , 6 , 9 – 14
Tuberculosis accounts for up to 70% of pericarditis in endemic areas but is a rare etiology in nonendemic areas. 9 – 14 , 16
Diagnosis of pericarditis requires at least two of the following criteria: new or worsening pericardial effusion, characteristic pleuritic chest pain, pericardial friction rub, or electrocardiographic (ECG) changes, including new, widespread ST elevations or PR depressions. 6 , 17
Pericarditis can be further classified as acute, incessant, recurrent, or chronic. 6
The differential diagnosis includes other causes of acute chest pain, such as thoracic artery aneurysm and dissection, acute coronary syndrome, mediastinitis, pulmonary embolism, pneumonia, pneumothorax, pneumopericardium, costochondritis, gastroesophageal reflux disease, neoplasm, or myocarditis ( Table 1 ) . 6 , 10 , 18 , 19
SIGNS AND SYMPTOMS
More than 90% of patients being evaluated for acute pericarditis report acute, retrosternal, pleuritic chest pain. Pain may radiate to the jaw, neck, or arms, but this should not be considered sensitive or specific for acute pericarditis. 6 , 20 , 21
Pain may be exacerbated by a supine position or improved by leaning forward. 6 In one cohort study, 46% of patients with pericarditis diagnosed in the emergency department experienced changes in pain with changes in posture. 21
Pain is not generally relieved by nitrates. 6 , 10 , 17
Patients with an infectious etiology of acute pericarditis may have fever, chills, myalgias, tachycardia, and leukocytosis. 6 , 22
Pericardial friction rubs are highly specific for pericarditis but have low sensitivity, with 18% to 84% of patients having a pericardial friction rub. 7 , 23 , 24 Friction rub is characterized by a transient, scratchy or squeaky quality that is best auscultated by the patient leaning forward and holding their breath. 6 , 10 , 17 , 21 An audible example of a friction rub is available.
Acute pericarditis can be complicated by cardiac tamponade, which is suggested by hypotension, pulsus paradoxus, increased jugular venous pressure, and a quiet precordium. 6
DIAGNOSTIC TESTING
Initial evaluation.
The initial evaluation should include patient history, physical examination, electrocardiography, chest radiography, transthoracic echocardiography, and baseline laboratory studies (i.e., complete blood count, basic metabolic panel, cardiac biomarkers, erythrocyte sedimentation rate, and serum C-reactive protein levels). 6
Classic ECG f indings include PR-segment depressions; diffuse, concave, upward ST-segment elevations without reciprocal changes; and T-wave inversions ( Figure 1 10 ) . These findings are common but are not present in all cases. 6 , 21
In contrast, ECG findings suggestive of ischemia or infarction include convex ST elevations that are regional without reciprocal ST depressions. PR depression is generally absent. 25 , 26
Elevation of C-reactive protein levels occurs in approximately 75% of patients suspected of having acute pericarditis; although it is not diagnostic, elevation can support the diagnosis. 27
In a retrospective observational study, an elevated neutrophil-lymphocyte ratio of 5.0 or greater was associated with an increased probability of recurrence of pericarditis and cardiac tamponade (odds ratio = 2.4; 95% CI, 1.7 to 3.4). 28
Cardiac biomarkers were elevated in 6.4% to 49% of patients in three observational studies. 21 , 29 , 30 In one cohort study, an increase in serum troponin levels was associated with one additional day of hospitalization. 31
Chest radiography can detect an enlarged cardiac silhouette, which supports the presence of pericardial effusion. 6
Transthoracic echocardiography is recommended for evaluation of patients with acute pericarditis because it can identify potential complications, such as tamponade or constrictive pericarditis. 10 , 18
Transthoracic echocardiography can indirectly quantify the size of pericardial effusion. Pericardial effusions larger than 21 mm on echocardiography are associated with a higher risk of complications. 18
In patients with suspected viral pericarditis, routine identification of the causative agent is not recommended unless hepatitis C or HIV infection is suspected. 17

Subsequent Testing for High-Risk or Hospitalized Patients
For patients who require hospitalization, evaluation should include obtaining blood and pericardial cultures to identify bacterial infections, genomic polymerase chain reaction testing to identify viruses, and antinuclear antibody testing to identify autoimmune conditions. 6 , 20
Computed tomography and cardiac magnetic resonance imaging are considered adjunct studies that could identify pericardial effusion or evidence of myocardial inflammation. 18 In a retrospective observational study, pericardial thickening or enhancement was the most accurate single parameter for pericarditis, with a sensitivity of 54% to 59% and a specificity of 91% to 96% (positive likelihood ratio = 8.1; negative likelihood ratio = 0.46). 32
Tuberculosis testing using interferon gamma release assay with acid-fast bacilli staining and cultures should be considered in areas where tuberculosis is endemic. 17 , 33
First-line treatment includes nonsteroidal anti-inflammatory drugs (NSAIDs), colchicine (Colcrys), and a proton pump inhibitor for gastro-protection. Ibuprofen and indomethacin are the most commonly used NSAIDs; aspirin is preferred in patients with comorbid coronary artery disease. 6 , 34
Expert opinion suggests that NSAIDs should be used for 7 to 10 days followed by a gradual taper once symptoms improve and C-reactive protein levels normalize. 27
Glucocorticoids are typically second-line therapy or used when NSAIDs are contraindicated (e.g., beyond 20 weeks' gestation, systemic inflammatory disease) due to a higher risk of recurrence and adverse effects. Prednisone dosages of 0.25 to 0.5 mg per kg per day followed by a slow taper minimize the risk of recurrence compared with higher dosages. 6
The addition of colchicine to first- or second-line agents significantly reduces the recurrence rate of acute pericarditis (absolute risk reduction = 22%; number needed to treat = 5) and recurrent pericarditis (absolute risk reduction = 23%; number needed to treat = 4). Colchicine should be used for 3 months in patients with acute pericarditis and at least 6 months in those with recurrent pericarditis. 35 – 38
Triple therapy with NSAIDs, glucocorticoids, and colchicine may be used in patients with recurrent pericarditis refractory to standard care, based on expert opinion and an observational study. If symptoms recur during glucocorticoid tapering, it is preferred to restart by increasing the dosage of the NSAID, not increasing the dosage of the glucocorticoid. 6 , 39 , 40
Use of anti–interleukin-1 agents, such as anakinra and rilonacept (Arcalyst), for the treatment of recurrent pericarditis refractory to standard treatment is supported by two small randomized controlled trials. A retrospective cohort study and a systematic review show weaker evidence for the use of azathioprine and intravenous immune globulin, respectively. 6 , 41 – 44
Pericardiocentesis may be performed for cardiac tamponade, for suspected bacterial or neoplastic etiology, or for symptomatic effusions that have not responded to standard therapy. 6
Major risk factors derived from multivariate analysis and minor risk factors derived from expert opinion are associated with a worse prognosis and should prompt consideration for hospital admission and further workup ( Figure 2 ) . 6 , 20
Based on expert opinion, athletes should not participate in competitive sports for 3 months after the diagnosis of acute pericarditis and 3 to 6 months if there is myopericarditis. It is reasonable to return to play in less time if serum biomarkers, left ventricular function, and ECG findings have normalized. 6 , 45

Mortality in acute pericarditis is low, with a rate of 1.1% in hospitalized patients. 2
Idiopathic pericarditis is generally self-limited and responds well to initial treatment within a few days. Less than 5% of patients experience poor symptom resolution with initial treatment. 6 , 35 , 37
Symptoms recur in 10% to 30% of patients despite an initial satisfactory response to treatment with an NSAID and colchicine. 35 , 37 , 46
Significant complications can include pericardial effusion causing cardiac tamponade, left ventricular dysfunction, arrhythmias, heart failure, and constrictive pericarditis. 6 , 47
Although patients with idiopathic recurrent pericarditis experience higher morbidity, serious late complications, including tamponade, left ventricular dysfunction, and constrictive pericarditis, are rare. 40 , 47 , 48
This article updates previous articles on this topic by Snyder, et al. 10 ; Tingle, et al. 49 ; Goyle and Walling 50 ; and Marinella . 51
Data Sources: A PubMed search was completed in Clinical Queries using the key term pericarditis. The search included meta-analyses, randomized controlled trials, clinical trials, and reviews. Essential Evidence Plus, POEMs, the Cochrane database, DynaMed, and UpToDate were also searched. Whenever possible, if studies used race and/or gender as patient categories but did not define how these categories were assigned, they were not included in our final review. If studies using these categories were determined to be essential and were therefore included, limitations were explicitly stated in the manuscript. Search dates: April, June, and November 2023, and March 2024.
The views expressed are those of the authors and do not reflect the official policy of the Madigan Army Medical Center, Uniformed Services University of the Health Sciences, U.S. Department of the Army, U.S. Department of Defense, or U.S. government.
Fruergaard P, Launbjerg J, Hesse B, et al. The diagnoses of patients admitted with acute chest pain but without myocardial infarction. Eur Heart J. 1996;17(7):1028-1034.
Kytö V, Sipilä J, Rautava P. Clinical profile and influences on outcomes in patients hospitalized for acute pericarditis. Circulation. 2014;130(18):1601-1606.
Gouriet F, Levy PY, Casalta JP, et al. Etiology of pericarditis in a prospective cohort of 1162 cases. Am J Med. 2015;128(7):784.e1-784.e8.
Kumar N, Pandey A, Jain P, et al. Acute pericarditis-associated hospitalization in the USA: a nationwide analysis, 2003–2012. Cardiology. 2016;135(1):27-35.
Vecchié A, Chiabrando JG, Dell MS, et al. Clinical presentation and outcomes of acute pericarditis in a large urban hospital in the United States of America. Chest. 2020;158(6):2556-2567.
Adler Y, Charron P, Imazio M, et al.; ESC Scientific Document Group. 2015 ESC guidelines for the diagnosis and management of pericardial diseases: the Task Force for the Diagnosis and Management of Pericardial Diseases of the European Society of Cardiology (ESC) endorsed by: the European Association for Cardio-Thoracic Surgery (EACTS). Eur Heart J. 2015;36(42):2921-2964.
Imazio M, Demichelis B, Parrini I, et al. Day-hospital treatment of acute pericarditis: a management program for outpatient therapy. J Am Coll Cardiol. 2004;43(6):1042-1046.
Imazio M, Cecchi E, Demichelis B, et al. Myopericarditis versus viral or idiopathic acute pericarditis. Heart. 2008;94(4):498-501.
Shakti D, Hehn R, Gauvreau K, et al. Idiopathic pericarditis and pericardial effusion in children: contemporary epidemiology and management. J Am Heart Assoc. 2014;3(6):e001483.
Snyder MJ, Bepko J, White M. Acute pericarditis: diagnosis and management. Am Fam Physician. 2014;89(7):553-560.
Zayas R, Anguita M, Torres F, et al. Incidence of specific etiology and role of methods for specific etiologic diagnosis of primary acute pericarditis. Am J Cardiol. 1995;75(5):378-382.
Levy PY, Fournier PE, Charrel R, et al. Molecular analysis of pericardial fluid: a 7-year experience. Eur Heart J. 2006;27(16):1942-1946.
Fallek Boldes O, Dahan S, Segal Y, et al. Characteristics of pericardial biopsy: 100 cases in a single center. Isr Med Assoc J. 2019;21(3):183-188.
Sathirareuangchai S, Kobayashi M, Shimizu D. Etiologies of pericarditis in hospital and forensic autopsies. Cardiovasc Pathol. 2020;49:107262.
Ariyarajah V, Spodick DH. Acute pericarditis: diagnostic cues and common electrocardiographic manifestations. Cardiol Rev. 2007;15(1):24-30.
Reuter H, Burgess LJ, Doubell AF. Epidemiology of pericardial effusions at a large academic hospital in South Africa. Epidemiol Infect. 2005;133(3):393-399.
Lazarou E, Tsioufis P, Vlachopoulos C, et al. Acute pericarditis: update. Curr Cardiol Rep. 2022;24(8):905-913.
Chiabrando JG, Bonaventura A, Vecchié A, et al. Management of acute and recurrent pericarditis: JACC state-of-the-art review. J Am Coll Cardiol. 2020;75(1):76-92.
Henzler T, Roeger S, Meyer M, et al. Pulmonary embolism: CT signs and cardiac biomarkers for predicting right ventricular dysfunction. Eur Respir J. 2012;39(4):919-926.
Imazio M, Spodick DH, Brucato A, et al. Controversial issues in the management of pericardial diseases. Circulation. 2010;121(7):916-928.
Hooper AJ, Celenza A. A descriptive analysis of patients with an emergency department diagnosis of acute pericarditis. Emerg Med J. 2013;30(12):1003-1008.
Radovanovic M, Petrovic M, Hanna RD, et al. Clinical presentation and management of methicillin-resistant Staphylococcus aureus pericarditis-systematic review. J Cardiovasc Dev Dis. 2022;9(4):103.
Spodick DH. Pericardial rub. Prospective, multiple observer investigation of pericardial friction in 100 patients. Am J Cardiol. 1975;35(3):357-362.
Prepoudis A, Koechlin L, Nestelberger T, et al.; APACE investigators. Incidence, clinical presentation, management, and outcome of acute pericarditis and myopericarditis. Eur Heart J Acute Cardiovasc Care. 2022;11(2):137-147.
Spodick DH. Diagnostic electrocardiographic sequences in acute pericarditis. Significance of PR segment and PR vector changes. Circulation. 1973;48(3):575-580.
Ginzton LE, Laks MM. The differential diagnosis of acute pericarditis from the normal variant: new electrocardiographic criteria. Circulation. 1982;65(5):1004-1009.
Imazio M, Brucato A, Maestroni S, et al. Prevalence of C-reactive protein elevation and time course of normalization in acute pericarditis: implications for the diagnosis, therapy, and prognosis of pericarditis. Circulation. 2011;123(10):1092-1097.
Yılmaz F, Yılmaz FK, Karagöz A, et al. Usefulness of neutrophil-to-lymphocyte ratio for predicting acute pericarditis outcomes. Acta Cardiol. 2022;77(5):422-430.
Imazio M, Demichelis B, Cecchi E, et al. Cardiac troponin I in acute pericarditis. J Am Coll Cardiol. 2003;42(12):2144-2148.
Bonnefoy E, Godon P, Kirkorian G, et al. Serum cardiac troponin I and ST-segment elevation in patients with acute pericarditis. Eur Heart J. 2000;21(10):832-836.
Machado S, Roubille F, Gahide G, et al. Can troponin elevation predict worse prognosis in patients with acute pericarditis?. Ann Cardiol Angeiol (Paris). 2010;59(1):1-7.
Hammer MM, Raptis CA, Javidan-Nejad C, et al. Accuracy of computed tomography findings in acute pericarditis. Acta Radiol. 2014;55(10):1197-1202.
Cremer PC, Kumar A, Kontzias A, et al. Complicated pericarditis: understanding risk factors and pathophysiology to inform imaging and treatment. J Am Coll Cardiol. 2016;68(21):2311-2328.
Lexicomp. Wolters Kluwer; 2023. Accessed May 4, 2023. https://www.wolterskluwer.com/en/solutions/lexicomp
Imazio M, Brucato A, Belli R, et al. Colchicine for the prevention of pericarditis: what we know and what we do not know in 2014–systematic review and meta-analysis. J Cardiovasc Med (Hagerstown). 2014;15(12):840-846.
Alabed S, Cabello JB, Irving GJ, et al. Colchicine for pericarditis. Cochrane Database Syst Rev. 2014(8):CD010652.
Imazio M, Belli R, Brucato A, et al. Efficacy and safety of colchicine for treatment of multiple recurrences of pericarditis (CORP-2): a multicentre, double-blind, placebo-controlled, randomised trial. Lancet. 2014;383(9936):2232-2237.
Imazio M, Brucato A, Forno D, et al. Efficacy and safety of colchicine for pericarditis prevention. Systematic review and meta-analysis. Heart. 2012;98(14):1078-1082.
Imazio M, Lazaros G, Brucato A, et al. Recurrent pericarditis: new and emerging therapeutic options. Nat Rev Cardiol. 2016;13(2):99-105.
Brucato A, Brambilla G, Moreo A, et al. Long-term outcomes in difficult-to-treat patients with recurrent pericarditis. Am J Cardiol. 2006;98(2):267-271.
Brucato A, Imazio M, Gattorno M, et al. Effect of anakinra on recurrent pericarditis among patients with colchicine resistance and corticosteroid dependence: the AIRTRIP randomized clinical trial. JAMA. 2016;316(18):1906-1912.
Klein AL, Imazio M, Cremer P, et al.; RHAPSODY Investigators. Phase 3 trial of interleukin-1 trap rilonacept in recurrent pericarditis. N Engl J Med. 2021;384(1):31-41.
Vianello F, Cinetto F, Cavraro M, et al. Azathioprine in isolated recurrent pericarditis: a single centre experience. Int J Cardiol. 2011;147(3):477-478.
Imazio M, Lazaros G, Picardi E, et al. Intravenous human immunoglobulins for refractory recurrent pericarditis: a systematic review of all published cases. J Cardiovasc Med (Hagerstown). 2016;17(4):263-269.
Pelliccia A, Solberg EE, Papadakis M, et al. Recommendations for participation in competitive and leisure time sport in athletes with cardiomyopathies, myocarditis, and pericarditis: position statement of the Sport Cardiology Section of the European Association of Preventive Cardiology (EAPC). Eur Heart J. 2019;40(1):19-33.
Imazio M, Brucato A, Cemin R, et al.; ICAP Investigators. A randomized trial of colchicine for acute pericarditis. N Engl J Med. 2013;369(16):1522-1528.
Imazio M, Brucato A, Maestroni S, et al. Risk of constrictive pericarditis after acute pericarditis. Circulation. 2011;124(11):1270-1275.
Imazio M, Brucato A, Adler Y, et al. Prognosis of idiopathic recurrent pericarditis as determined from previously published reports. Am J Cardiol. 2007;100(6):1026-1028.
Tingle LE, Molina D, Calvert CW. Acute pericarditis. Am Fam Physician. 2007;76(10):1509-1514.
Goyle KK, Walling AD. Diagnosing pericarditis. Am Fam Physician. 2002;66(9):1695-1702.
Marinella MA. Electrocardiographic manifestations and differential diagnosis of acute pericarditis. Am Fam Physician. 1998;57(4):699-704.
Continue Reading

More in AFP
Copyright © 2024 by the American Academy of Family Physicians.
This content is owned by the AAFP. A person viewing it online may make one printout of the material and may use that printout only for his or her personal, non-commercial reference. This material may not otherwise be downloaded, copied, printed, stored, transmitted or reproduced in any medium, whether now known or later invented, except as authorized in writing by the AAFP. See permissions for copyright questions and/or permission requests.
Copyright © 2024 American Academy of Family Physicians. All Rights Reserved.
IMAGES
VIDEO
COMMENTS
Review the typical presentation of a patient with pulmonary hypertension. ... Pulmonary hypertension associated with congenital heart disease ... (<1 month), known right atrial/ventricular thrombus, mechanical right heart valve, tricuspid valve clip, and acute infection. The most feared complication of the procedure is the rupture of the PA.
Pulmonary hypertension (PH) is classified into 5 clinical subgroups: pulmonary arterial hypertension (PAH), PH due to left-sided heart disease, PH due to chronic lung disease, chronic thromboembolic PH (CTEPH), and PH with an unclear and/or multifactorial mechanisms. A range of underlying conditions can lead to these disorders. Overall, PH affects approximately 1% of the global population, and ...
Pulmonary hypertension. When blood vessels in the lungs become thickened, narrowed, blocked or destroyed, it's harder for blood to flow through the lungs. As a result, blood pressure increases in the lungs. ... Pulmonary hypertension in 2021: Part I — definition, classification, pathophysiology, and presentation. Journal of Cardiothoracic and ...
Dyspnea on exertion is the most frequent presentation. Fatigue. (#2) ... qualitative signs of pulmonary hypertension may be seen. Chamber dilation: Dilated coronary sinus (although this may also be incidental, due to a persistent left superior vena cava, or due to congenital heart disease). ... dosing of IV epoprostenol for acute right heart ...
Pulmonary hypertension (PH) is a heterogeneous and highly morbid disease encountered commonly in general medicine, cardiology, and pulmonary medicine clinical practices. 1 The original definition of PH used mean pulmonary artery pressure (mPAP) ≥25 mm Hg, but this was derived from expert consensus opinion originally reported 45 years ago in the absence of sufficiently powered clinical data.
Patients suspected of having pulmonary hypertension (PH) undergo extensive diagnostic testing. The purpose of diagnostic testing is to confirm that PH exists and identify the underlying cause so that appropriate treatment can be administered. The clinical features, diagnostic evaluation, and diagnostic criteria for PH are reviewed here.
1 - 3. In patients with pulmonary hypertension due to lung disease or left heart disease, treatment should focus on optimizing comorbid conditions. C. 3, 9 - 11. In patients with pulmonary ...
2022 ESC/ERS pulmonary hypertension guidelines incorporate changes and adaptations focusing on clinical management https://bit.ly/3QtUvb4 ... Clinical presentation 23 5.1.2. Electrocardiogram 23 5.1.3. Chest radiography 24 ... Follow-up After Acute Pulmonary Embolism) showed a cumulative 2 year incidence of 2.3% and 16.0% for CTEPH and post-PE ...
Acute PAH is distinctive because they differ in their clinical presentation, diagnostic findings, and response to treatment from chronic PAH. The acute PAH may take either the form of acute onset of chronic PAH or acute PAH or surgery-related PAH. ... In acute pulmonary hypertension associated with decreased cardiac contractility and/or SVR ...
Classification of Pulmonary Hypertension. The modern classification for PH was established in 1998. 10 The intention of the classification was to group patients who appeared to share common mechanisms of disease. What emerged was a schema that classifies PH diagnoses into 5 distinct groups: PAH (Group 1); PH secondary to left heart disease ...
Pulmonary hypertension is hard to diagnose early because it's not often found during a routine physical exam. Even when pulmonary hypertension is more advanced, its symptoms are similar to those of other heart and lung conditions. ... Pulmonary hypertension in 2021: Part I — definition, classification, pathophysiology, and presentation ...
Pulmonary hypertension is classified into 5 groups. Suspect pulmonary hypertension if patients have dyspnea unexplained by another clinically evident cardiac or pulmonary disorder. Begin diagnostic testing with chest x-ray, spirometry, ECG, and transthoracic Doppler echocardiography. Confirm the diagnosis by right heart catheterization.
Pulmonary arterial hypertension (PAH), the first category of pulmonary hypertension, is a chronic and progressive disorder characterised by angioproliferative vasculopathy in the pulmonary arterioles, leading to endothelial and smooth muscle proliferation and dysfunction, inflammation and thrombosis. These changes increase pulmonary vascular ...
These comprehensive clinical practice guidelines cover the whole spectrum of PH with an emphasis on diagnosing and treating pulmonary arterial hypertension (PAH) and chronic thrombo-embolic pulmonary hypertension (CTEPH). Guidelines and related materials are for use by individuals for personal or educational purposes. No commercial use is allowed.
What is pulmonary hypertension? Pulmonary hypertension happens when the pressure in the blood vessels leading from the heart to the lungs is too high. With pulmonary hypertension, the blood vessels to the lungs develop an increased amount of muscle in the wall of the blood vessels. The heart pumps blood from the right ventricle to the lungs to ...
Guidelines summarize and evaluate available evidence with the aim of assisting health professionals in proposing the best management strategies for an individual patient with a given condition. Guidelines and their recommendations should facilitate decision making of health professionals in their daily practice. However, the final decisions concerning an individual patient must be made by the ...
Key Points:-Myriad of chief complaints can be pulmonary hypertension; try to determine what is triggering the acute decompensation. -Elevations in troponin and liver function tests portend a poor prognosis. -Point of care ultrasound may be useful in guiding acute resuscitation, though evaluation of IVC may not reflect intravascular volume. -Avoid hypoxemia and hypercarbia and maintain ...
Pulmonary hypertension (PH) is a heterogeneous group of disorders that result in an elevation in blood pressure in the pulmonary arteries (mean pulmonary artery [PA] pressure [mPAP] >20 mm Hg). 1 Increased mPAP is consistently associated with an increased risk of death. 2 The prevalence of PH is increasing, with an aging population, a rising prevalence of heart and lung disease, and improved ...
Patients with pulmonary hypertension (PH) can be extremely challenging to manage in the critical care setting. In this article we review the classification, diagnosis, and chronic management of PH. An approach to the management of the critically unwell PH patient is discussed. Initial management involves treating underlying precipitants of deterioration and optimizing right ventricular (RV ...
The field of pulmonary hypertension has been transformed beyond recognition over the past 30 years. Advances in the understanding of pathophysiology has led to the testing of multiple therapies and interventions in clinical trials. This productive bench-to-bedside pipeline, from basic mechanisms through to evidence-based clinical guidelines, is something to be celebrated.
Acute pulmonary arterial hypertension (PAH), which may complicate the course of many complex disorders, is always underdiagnosed and its treatment frequently begins only after serious complications have developed. Acute PAH is distinctive because they differ in their clinical presentation, diagnosti …
The clinical presentation of a patient in acute decompensated pulmonary hypertension can often be falsely reassuring at rest. Although systemic arterial pressure at admission appeared to be a prognostic factor [ 21 ], a majority of patients are able to maintain a normal level of systemic vascular resistance for a long time.
Identification of long-term calcium channel blocker (CCB) responders with acute vasodilator challenge is critical in the evaluation of patients with pulmonary arterial hypertension. Currently there is no standardized approach for use of supplemental oxygen during acute vasodilator challenge. In this …
Ace the Case: Pulmonary Arterial Hypertension (PAH) Presentation and ...
A Critical Confluence: Acute Pulmonary Embolism Complicating Systemic Sclerosis-Associated Pulmonary Arterial Hypertension @article{Lam2024ACC, title={A Critical Confluence: Acute Pulmonary Embolism Complicating Systemic Sclerosis-Associated Pulmonary Arterial Hypertension}, author={CM Lam and M. Kioka}, journal={A60.
Pulmonary arterial hypertension: Infectious Bacterial Actinomyces neuii ... Dell MS, et al. Clinical presentation and outcomes of acute pericarditis in a large urban hospital in the United States ...
Diagnostic and therapeutic decision-making in pregnancy with suspected pulmonary embolism (PE) is challenging. European and other international professional societies have proposed various recommendations that are ambiguous, probably due to the unavailability of randomized controlled trials. In the following sections, we discuss the supporting diagnostic steps and treatments. We suggest a ...
DOI: 10.1164/ajrccm-conference.2024.209.1_meetingabstracts.a2217 Corpus ID: 269492233; Acute Pulmonary Hypertension Caused by Uterine Artery Embolization for Placenta Accreta @article{Tiwari2024AcutePH, title={Acute Pulmonary Hypertension Caused by Uterine Artery Embolization for Placenta Accreta}, author={M. Tiwari and N. Rupani and A. Kassem and I. El Husseini}, journal={A60.
@article{Tachibana2024SquamousMI, title={Squamous metaplasia is an indicator of acute exacerbation in patients with usual interstitial pneumonia / idiopathic pulmonary fibrosis.}, author={Yuri Tachibana and Masatake Hara and Mikiko Hashisako and Yasuhiko Yamano and Kensuke Kataoka and Yasuhiro Kondoh and Takeshi Johkoh and Shimpei Morimoto and ...
Several cardiac risk factors are commonly observed in individuals with acute coronary syndrome (ACS). Traditional risk factors for coronary artery disease include smoking, hypertension, diabetes, hyperlipidaemia, and a hereditary predisposition to the condition. 1 Young people facing myocardial infarction (MI) encounter significant challenges as the disease is less likely caused by ...