
BiteSized Immunology: Systems & Processes

Antigen Processing and Presentation
In order to be capable of engaging the key elements of adaptive immunity (specificity, memory, diversity, self/nonself discrimination), antigens have to be processed and presented to immune cells. Antigen presentation is mediated by MHC class I molecules , and the class II molecules found on the surface of antigen-presenting cells (APCs) and certain other cells.
MHC class I and class II molecules are similar in function: they deliver short peptides to the cell surface allowing these peptides to be recognised by CD8+ (cytotoxic) and CD4+ (helper) T cells, respectively. The difference is that the peptides originate from different sources – endogenous, or intracellular , for MHC class I; and exogenous, or extracellular for MHC class II. There is also so called cross-presentation in which exogenous antigens can be presented by MHC class I molecules. Endogenous antigens can also be presented by MHC class II when they are degraded through autophagy.

MHC class I presentation
MHC class I molecules are expressed by all nucleated cells. MHC class I molecules are assembled in the endoplasmic reticulum (ER) and consist of two types of chain – a polymorphic heavy chain and a chain called β2-microglobulin. The heavy chain is stabilised by the chaperone calnexin , prior to association with the β2-microglobulin. Without peptides, these molecules are stabilised by chaperone proteins : calreticulin, Erp57, protein disulfide isomerase (PDI) and tapasin. The complex of TAP, tapasin, MHC class I, ERp57 and calreticulin is called the peptide-loading complex (PLC). Tapasin interacts with the transport protein TAP (transporter associated with antigen presentation) which translocates peptides from the cytoplasm into the ER. Prior to entering the ER, peptides are derived from the degradation of proteins, which can be of viral- or self origin. Degradation of proteins is mediated by cytosolic- and nuclear proteasomes, and the resulting peptides are translocated into the ER by means of TAP. TAP translocates peptides of 8 –16 amino acids and they may require additional trimming in the ER before binding to MHC class I molecules. This is possibly due to the presence of ER aminopeptidase (ERAAP) associated with antigen processing.
It should be noted that 30–70% of proteins are immediately degraded after synthesis (they are called DRiPs – defective ribosomal products, and they are the result of defective transcription or translation). This process allows viral peptides to be presented very quickly – for example, influenza virus can be recognised by T cells approximately 1.5 hours post-infection. When peptides bind to MHC class I molecules, the chaperones are released and peptide–MHC class I complexes leave the ER for presentation at the cell surface. In some cases, peptides fail to associate with MHC class I and they have to be returned to the cytosol for degradation. Some MHC class I molecules never bind peptides and they are also degraded by the ER-associated protein degradation (ERAD) system.
There are different proteasomes that generate peptides for MHC class-I presentation: 26S proteasome , which is expressed by most cells; the immunoproteasome, which is expressed by many immune cells; and the thymic-specific proteasome expressed by thymic epithelial cells.
Antigen presentation
On the surface of a single cell, MHC class I molecules provide a readout of the expression level of up to 10,000 proteins. This array is interpreted by cytotoxic T lymphocytes and Natural Killer cells, allowing them to monitor the events inside the cell and detect infection and tumorigenesis.
MHC class I complexes at the cell surface may dissociate as time passes and the heavy chain can be internalised. When MHC class I molecules are internalised into the endosome, they enter the MHC class-II presentation pathway. Some of the MHC class I molecules can be recycled and present endosomal peptides as a part of a process which is called cross-presentation .
The usual process of antigen presentation through the MHC I molecule is based on an interaction between the T-cell receptor and a peptide bound to the MHC class I molecule. There is also an interaction between the CD8+ molecule on the surface of the T cell and non-peptide binding regions on the MHC class I molecule. Thus, peptide presented in complex with MHC class I can only be recognised by CD8+ T cells. This interaction is a part of so-called ‘three-signal activation model’, and actually represents the first signal. The next signal is the interaction between CD80/86 on the APC and CD28 on the surface of the T cell, followed by a third signal – the production of cytokines by the APC which fully activates the T cell to provide a specific response.
MHC class I polymorphism
Human MHC class I molecules are encoded by a series of genes – HLA-A, HLA-B and HLA-C (HLA stands for ‘Human Leukocyte Antigen’, which is the human equivalent of MHC molecules found in most vertebrates). These genes are highly polymorphic, which means that each individual has his/her own HLA allele set. The consequences of these polymorphisms are differential susceptibilities to infection and autoimmune diseases that may result from the high diversity of peptides that can bind to MHC class I in different individuals. Also, MHC class I polymorphisms make it virtually impossible to have a perfect tissue match between donor and recipient, and thus are responsible for graft rejection.

MHC class II presentation
MHC class II molecules are expressed by APCs, such as dendritic cells (DC), macrophages and B cells (and, under IFNγ stimuli, by mesenchymal stromal cells, fibroblasts and endothelial cells, as well as by epithelial cells and enteric glial cells). MHC class II molecules bind to peptides that are derived from proteins degraded in the endocytic pathway. MHC class II complexes consists of α- and β-chains that are assembled in the ER and are stabilised by invariant chain (Ii). The complex of MHC class II and Ii is transported through the Golgi into a compartment which is termed the MHC class II compartment (MIIC). Due to acidic pH, proteases cathepsin S and cathepsin L are activated and digest Ii, leaving a residual class II-associated Ii peptide (CLIP) in the peptide-binding groove of the MHC class II. Later, the CLIP is exchanged for an antigenic peptide derived from a protein degraded in the endosomal pathway. This process requires the chaperone HLA-DM, and, in the case of B cells, the HLA-DO molecule. MHC class II molecules loaded with foreign peptide are then transported to the cell membrane to present their cargo to CD4+ T cells. Thereafter, the process of antigen presentation by means of MHC class II molecules basically follows the same pattern as for MHC class I presentation.
As opposed to MHC class I, MHC class II molecules do not dissociate at the plasma membrane. The mechanisms that control MHC class II degradation have not been established yet, but MHC class II molecules can be ubiquitinised and then internalised in an endocytic pathway.
MHC class II polymorphism
Like the MHC class I heavy chain, human MHC class II molecules are encoded by three polymorphic genes: HLA-DR, HLA-DQ and HLA-DP. Different MHC class II alleles can be used as genetic markers for several autoimmune diseases, possibly owing to the peptides that they present.

- DNA Replication
- Active Transport
- Cellular Receptors
- Endocytosis and Exocytosis
- Enzyme Inhibition
- Enzyme Kinetics
- Protein Structure
- Transcription of DNA
- Translation of DNA
- Anaerobic Respiration
- Electron Transport Chain
- Gluconeogenesis
- Calcium Regulation
- External Balance of Potassium
- Internal Balance of Potassium
- Sodium Regulation
- Cell Membrane
- Endoplasmic Reticulum
- Golgi Apparatus
- Mitochondria
- Blood Vessels
- Cellular Adaptations
- Epithelial Cells
- Muscle Histology
- Structure of Glands
- Control of Stroke Volume
- Control of Heart Rate
- Cardiac Cycle
- Cardiac Pacemaker Cells
- Conduction System
- Contraction of Cardiac Muscle
- Ventricular Action Potentials
- Blood Flow in Vessels
- Control of Blood Pressure
- Capillary Exchange
- Flow In Peripheral Circulation
- Venous Return
- Cardiac Muscle
- Hepatic Circulation
- Skeletal Muscle
- Airway Resistance
- Lung Volumes
- Mechanics of Breathing
- Gas Exchange
- Oxygen Transport in The Blood
- Transport of Carbon Dioxide in the Blood
- Ventilation-Perfusion Matching
- Chemoreceptors
- Cough Reflex
- Neural Control of Ventilation
- Respiratory Regulation of Acid-Base Balance
- Responses of The Respiratory System to Stress
- Regulation of Saliva
- Secretion of Saliva
- Gastric Acid Production
- Gastric Mucus Production
- Digestion and Absorption
- Histology and Cellular Function of the Small Intestine
- Absorption in the Large Intestine
- Large Intestinal Motility
- Bilirubin Metabolism
- Carbohydrate Metabolism in the Liver
- Lipid Metabolism in the Liver
- Protein and Ammonia Metabolism in the Liver
- Storage Functions of the Liver
- Bile Production
- Function of The Spleen
- Exocrine Pancreas
- Somatostatin
- Proximal Convoluted Tubule
- Loop of Henle
- Distal Convoluted Tubule and Collecting Duct
- Storage Phase of Micturition
- Voiding Phase of Micturition
- Antidiuretic Hormone
- Renin-Angiotensin-Aldosterone System
- Urinary Regulation of Acid-Base Balance
- Water Filtration and Reabsorption
- Development of the Reproductive System
- Gametogenesis
- Gonadotropins and the Hypothalamic Pituitary Axis
- Menstrual Cycle
- Placental Development
- Fetal Circulation
- Maternal Adaptations in Pregnancy
- Cells of the Nervous System
- Central Nervous System
- Cerebrospinal Fluid
- Neurotransmitters
- Peripheral Nervous System
- Action Potential
- Excitatory and Inhibitory Synaptic Signalling
- Resting Membrane Potential
- Synaptic Plasticity
- Synaptic Transmission
- Ascending Tracts
- Auditory Pathway
- Consciousness and Sleep
- Modalities of Sensation
- Pain Pathways
- Sensory Acuity
- Visual Pathway
- Descending Tracts
- Lower Motor Neurones
- Muscle Stretch Reflex
- Upper Motor Neurones
- Aqueous Humour
- Ocular Accommodation
- Thyroid Gland
- Parathyroid Glands
- Adrenal Medulla
- Zona Glomerulosa
- Zona Fasciculata
- Zona Reticularis
- Endocrine Pancreas
- The Hypothalamus
- Anterior Pituitary
- Posterior Pituitary
- White Blood Cells – Summary
- Barriers to Infection
- Infection Recognition Molecules
- Phagocytosis
- The Complement System
Antigen Processing and Presentation
- Primary and Secondary Immune Responses
- T Cell Memory
- Acute Inflammation
- Autoimmunity
- Chronic Inflammation
- Hypersensitivity Reactions
- Immunodeficiency
- Types of Immunity
- Antibiotics
- Viral Infection
- Blood Groups
- Coagulation
- Erythropoiesis
- Iron Metabolism
- Mononuclear Phagocyte System
Original Author(s): Antonia Round Last updated: 17th July 2023 Revisions: 9
- 1 Antigen Presentation
- 2.1 MHC Class I Molecules
- 2.2 MCH Class II Molecules
- 3.1 T Cell Receptors
- 3.2 Co-Receptors
- 4 Clinical Relevance – Autoimmune disease
T cells can only recognise antigens when they are displayed on cell surfaces. This is carried out by Antigen-presenting cells (APCs) , the most important of which are dendritic cells, B cells, and macrophages. APCs can digest proteins they encounter and display peptide fragments from them on their surfaces for other immune cells to recognise.
This process of antigen presentation allows T cells to “see” what proteins are present in the body and to form an adaptive immune response against them. In this article, we shall discuss antigen processing, presentation, and recognition by T cells.
Antigen Presentation
Antigens are delivered to the surface of APCs by Major Histocompatibility Complex (MHC) molecules. Different MHC molecules can bind different peptides. The MHC is highly polygenic and polymorphic which equips us to recognise a vast array of different antigens we might encounter. There are different classes of MHC, which have different functions:
- MHC class I molecules are found on all nucleated cells (not just professional APCs) and typically present intracellular antigens such as viruses.
- MHC class II molecules are only found on APCs and typically present extracellular antigens such as bacteria.
This is logical because should a virus be inside a cell of any type, the immune system needs to be able to respond to it. This also explains why pathogens inside human red blood cells (which are non-nucleated) can be difficult for the immune system to find, such as in malaria.
Whilst this is the general rule, in cross-presentation extracellular antigens can be presented by MHC class I, and in autophagy intracellular antigens can be presented by MHC class II.
Antigen Processing
Before an antigen can be presented, it must first be processed . Processing transforms proteins into antigenic peptides.
MHC Class I Molecules
Intracellular peptides for MHC class I presentation are made by proteases and the proteasome in the cytosol, then transported into the endoplasmic reticulum via TAP (Transporter associated with Antigen Processing) to be further processed.
They are then assembled together with MHC I molecules and travel to the cell surface ready for presentation.
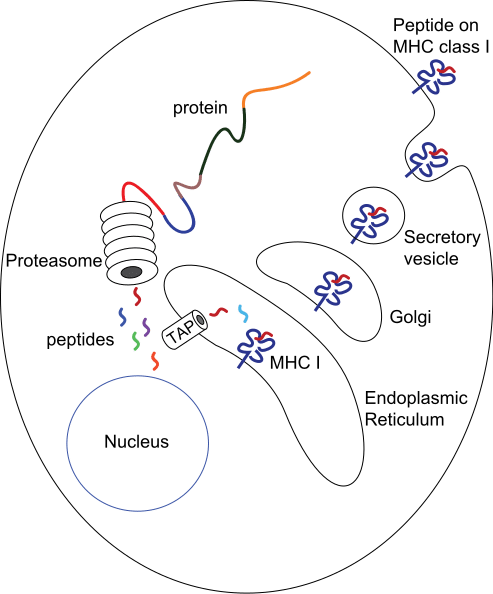
Fig 1 – Diagram demonstrating the production of peptides for MHC class I presentation
MCH Class II Molecules
The route of processing for exogenous antigens for MHC class II presentation begins with endocytosis of the antigen. Once inside the cell, they are encased within endosomes that acidify and activate proteases, to degrade the antigen.
MHC class II molecules are transported into endocytic vesicles where they bind peptide antigen and then travel to the cell surface.
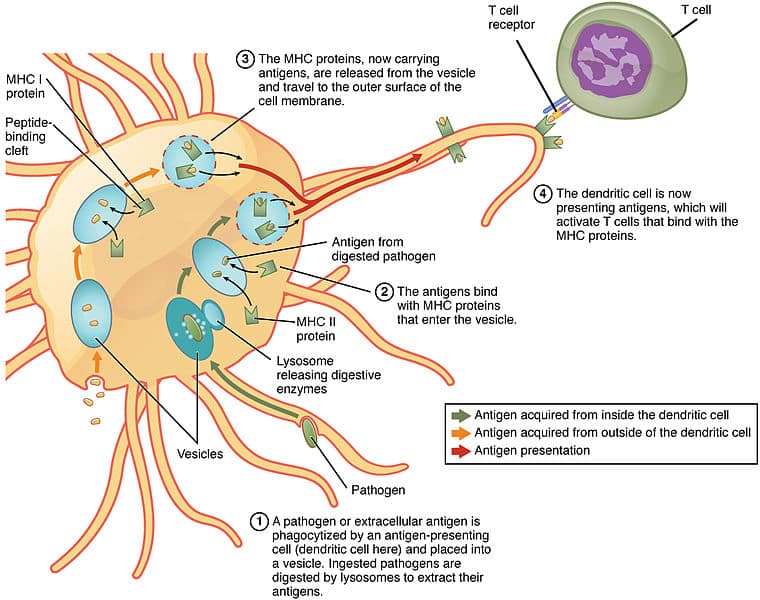
Fig 2 – Diagram showing processing of antigens for MHC Class II presentation by a dendritic cell
The antigen presented on MHCs is recognised by T cells using a T cell receptor (TCR) . These are antigen-specific .
T Cell Receptors
Each T cell has thousands of TCRs , each with a unique specificity that collectively allows our immune system to recognise a wide array of antigens.
This diversity in TCRs is achieved through a process called V(D)J recombination during development in the thymus. TCR chains have a variable region where gene segments are randomly rearranged, using the proteins RAG1 and RAG2 to initiate cleavage and non-homologous end joining to rejoin the chains.
The diversity of the TCRs can be further increased by inserting or deleting nucleotides at the junctions of gene segments; together forming the potential to create up to 10 15 unique TCRs.
TCRs are specific not only for a particular antigen but also for a specific MHC molecule. T cells will only recognise an antigen if a specific antigen with a specific MHC molecule is present: this phenomenon is called MHC restriction .
Co-Receptors
As well as the TCR, another T cell molecule is required for antigen recognition and is known as a co-receptor. These are either a CD4 or CD8 molecule:
- CD4 is present on T helper cells and only binds to antigen-MHC II complexes.
- CD8 is present on cytotoxic T cells and only binds to antigen-MHC I complexes.
This, therefore, leads to very different effects. Antigens presented with MHC II will activate T helper cells and antigens presented with MHC I activate cytotoxic T cells. Cytotoxic T cells will kill the cells that they recognise, whereas T helper cells have a broader range of effects on the presenting cell such as activation to produce antibodies (in the case of B cells) or activation of macrophages to kill their intracellular pathogens.
Clinical Relevance – Autoimmune disease
It is important to note that APCs may deliver foreign antigens or self-antigens. In the case of autoimmune diseases, self-antigens are presented to T cells, which then initiates an immune response against our own tissues.
For example, in Graves’ disease , TSHR (thyroid stimulating hormone receptor) acts as a self-antigen and is presented to T cells. This then activates B cells to produce autoantibodies against TSHRs in the thyroid. This results in the activation of TSHRs leading to hyperthyroidism and a possible goitre.
[start-clinical]
Clinical Relevance - Autoimmune disease
[end-clinical]
Found an error? Is our article missing some key information? Make the changes yourself here!
Once you've finished editing, click 'Submit for Review', and your changes will be reviewed by our team before publishing on the site.
We use cookies to improve your experience on our site and to show you relevant advertising. To find out more, read our privacy policy .
Privacy Overview

- school Campus Bookshelves
- menu_book Bookshelves
- perm_media Learning Objects
- login Login
- how_to_reg Request Instructor Account
- hub Instructor Commons
- Download Page (PDF)
- Download Full Book (PDF)
- Periodic Table
- Physics Constants
- Scientific Calculator
- Reference & Cite
- Tools expand_more
- Readability
selected template will load here
This action is not available.
20.3E: Antigen-Presenting Cells
- Last updated
- Save as PDF
- Page ID 7949
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
Antigen presentation is a process by which immune cells capture antigens and then enable their recognition by T cells.
Learning Objectives
- Describe the role of antigen-presenting cells
- The host’s cells express “self” antigens that identify them as such. These antigens are different from those in bacteria (“non-self” antigens) and in virus-infected host cells (“missing-self”).
- Antigen presentation consists of pathogen recognition, phagocytosis of the pathogen or its molecular components, processing of the antigen, and then presentation of the antigen to naive T cells.
- The T cell receptor is restricted to recognizing antigenic peptides only when bound to appropriate molecules of the major histocompatibility complex (MHC), also known in humans as human leukocyte antigen (HLA).
- Helper T cells recieve antigens from MHC II on an APC, while cytotoxic T cells recieve antigens from MHC I. Helper T cells present their antigen to B cells as well.Dendritic cells, B cells, and macrophages play a major role in the innate response, and are the primary antigen-presenting cells (APC).
- APCs use toll-like receptors to identify PAMPS and DAMPs, which are signs of an infection and may be processed into antigen peptides if phagocytized. Most APCs cannot tell the difference between different types of antigens like B and T cells can.
- damage-associated molecular pattern : Protein or nucleic acid based signs of pathogen induced damage. Protein DAMPs may be phagocytized and processed for antigen presentation.
- cytotoxic : A population of T cells specialized for inducing the deaths of other cells.
Antigen presentation is a process in the body’s immune system by which macrophages, dendritic cells and other cell types capture antigens, then present them to naive T-cells. The basis of adaptive immunity lies in the capacity of immune cells to distinguish between the body’s own cells and infectious pathogens. The host’s cells express “self” antigens that identify them as belonging to the self. These antigens are different from those in bacteria (“non-self” antigens) or in virally-infected host cells (“missing-self”). Antigen presentation broadly consists of pathogen recognition, phagocytosis of the pathogen or its molecular components, processing of the antigen, and then presentation of the antigen to naive (mature but not yet activated) T cells. The ability of the adaptive immune system to fight off pathogens and end an infection depends on antigen presentation.
Antigen Presenting Cells
Antigen Presenting Cells (APCs) are cells that capture antigens from within the body, and present them to naive T-cells. Many immune system cells can present antigens, but the most common types are macrophages and dendritic cells, which are two types of terminally differentiated leukocytes that arise from monocytes. Both of these APCs perform many immune functions that are important for both innate and adaptive immunity, such as removing leftover pathogens and dead neutrophils after an inflammatory response. Dendritic cells (DCs) are generally found in tissues that have contact with the external environment (such as the skin or respiratory epithelium) while macrophages are found in almost all tissues. Some types of B cells may also present antigens as well, though it is not their primary function.
APCs phagocytize exogenous pathogens such as bacteria, parasites, and toxins in the tissues and then migrate, via chemokine signals, to lymph nodes that contain naive T cells. During migration, APCs undergo a process of maturation in which they digest phagocytized pathogens and begin to express the antigen in the form of a peptide on their MHC complexes, which enables them to present the antigen to naive T cells. The antigen digestion phase is also called “antigen processing,” because it prepares the antigens for presentation. This MHC:antigen complex is then recognized by T cells passing through the lymph node. Exogenous antigens are usually displayed on MHC Class II molecules, which interact with CD4+ helper T cells.
This maturation process is dependent on signaling from other pathogen-associated molecular pattern (PAMP) molecules (such as a toxin or component of a cell membrane from a pathogen) through pattern recognition receptors (PRRs), which are received by Toll-like receptors on the DC’s body. They may also recognize damage-associated molecular pattern (DAMP) molecules, which include degraded proteins or nucleic acids released from cells that undergo necrosis. PAMPs and DAMPS are not technically considered antigens themselves, but instead are signs of pathogen presence that alert APCs through Toll-like receptor binding. However if a DC phagocytzes a PAMP or DAMP, it could be used as an antigen during antigen presentation. APCs are unable to distinguish between different types of antigens themselves, but B and T cells can due to their specificity.
Antigen Presentation
T cells must be presented with antigens in order to perform immune system functions. The T cell receptor is restricted to recognizing antigenic peptides only when bound to appropriate molecules of the MHC complexes on APCs, also known in humans as Human leukocyte antigen (HLA).
Several different types of T cell can be activated by APCs, and each type of T cell is specially equipped to deal with different pathogens, whether the pathogen is bacterial, viral or a toxin. The type of T cell activated, and therefore the type of response generated, depends on which MHC complex the processed antigen-peptide binds to.
MHC Class I molecules present antigen to CD8+ cytotoxic T cells, while MHC class II molecules present antigen to CD4+ helper T cells. With the exception of some cell types (such as erythrocytes), Class I MHC is expressed by almost all host cells. Cytotoxic T cells (also known as TC, killer T cell, or cytotoxic T-lymphocyte (CTL)) are a population of T cells that are specialized for inducing the death of other cells. Recognition of antigenic peptides through Class I by CTLs leads to the killing of the target cell, which is infected by virus, intracytoplasmic bacterium, or are otherwise damaged or dysfunctional. Additionally, some helper T cells will present their antigen to B cells, which will activate their proliferation response.
Antigen presentation : In the upper pathway; foreign protein or antigen (1) is taken up by an antigen-presenting cell (2). The antigen is processed and displayed on a MHC II molecule (3), which interacts with a T helper cell (4). In the lower pathway; whole foreign proteins are bound by membrane antibodies (5) and presented to B lymphocytes (6), which process (7) and present antigen on MHC II (8) to a previously activated T helper cell (10), spurring the production of antigen-specific antibodies (9).
LICENSES AND ATTRIBUTIONS
CC LICENSED CONTENT, SHARED PREVIOUSLY
- Curation and Revision. Authored by : Boundless.com. Provided by : Boundless.com. License : CC BY-SA: Attribution-ShareAlike
CC LICENSED CONTENT, SPECIFIC ATTRIBUTION
- antigen. Provided by : Wiktionary. Located at : en.wiktionary.org/wiki/antigen . License : CC BY-SA: Attribution-ShareAlike
- Adaptive immune system. Provided by : Wikipedia. Located at : en.Wikipedia.org/wiki/Adaptive_immune_system . License : CC BY-SA: Attribution-ShareAlike
- antibody. Provided by : Wiktionary. Located at : en.wiktionary.org/wiki/antibody . License : CC BY-SA: Attribution-ShareAlike
- macrophage. Provided by : Wiktionary. Located at : en.wiktionary.org/wiki/macrophage . License : CC BY-SA: Attribution-ShareAlike
- Antibody. Provided by : Wikipedia. Located at : en.Wikipedia.org/wiki/File:Antibody.jpg . License : Public Domain: No Known Copyright
- Antigen presentation. Provided by : Wikipedia. Located at : en.Wikipedia.org/wiki/File:Antigen_presentation.svg . License : Public Domain: No Known Copyright
- Immune system. Provided by : Wikipedia. Located at : en.Wikipedia.org/wiki/Immune_system%23Innate_immune_system . License : CC BY-SA: Attribution-ShareAlike
- Lymphocyte_activation_simple.png. Provided by : Wikipedia. Located at : en.Wikipedia.org/wiki/Immune_system#/media/File:Lymphocyte_activation_simple.png . License : CC BY-SA: Attribution-ShareAlike
- T cell. Provided by : Wikipedia. Located at : en.Wikipedia.org/wiki/T_cell . License : CC BY-SA: Attribution-ShareAlike
- B cell. Provided by : Wikipedia. Located at : en.Wikipedia.org/wiki/B_cell . License : CC BY-SA: Attribution-ShareAlike
- Immune cells. Provided by : Wikipedia. Located at : en.Wikipedia.org/wiki/Immune_cells . License : CC BY-SA: Attribution-ShareAlike
- thymus. Provided by : Wiktionary. Located at : en.wiktionary.org/wiki/thymus . License : CC BY-SA: Attribution-ShareAlike
- Red White Blood cells. Provided by : Wikipedia. Located at : en.Wikipedia.org/wiki/File:Red_White_Blood_cells.jpg . License : Public Domain: No Known Copyright
- Lymphocyte. Provided by : Wikipedia. Located at : en.Wikipedia.org/wiki/Lymphocyte . License : CC BY-SA: Attribution-ShareAlike
- B cells. Provided by : Wikipedia. Located at : en.Wikipedia.org/wiki/B%20cells . License : CC BY-SA: Attribution-ShareAlike
- natural killer (NK) cells. Provided by : Wikipedia. Located at : en.Wikipedia.org/wiki/natural...20(NK)%20cells . License : CC BY-SA: Attribution-ShareAlike
- T cells. Provided by : Wiktionary. Located at : en.wiktionary.org/wiki/T_cells . License : CC BY-SA: Attribution-ShareAlike
- Lymphocyte. Provided by : Wikipedia. Located at : en.Wikipedia.org/wiki/Lymphocyte . License : Public Domain: No Known Copyright
- Antigen presentation. Provided by : Wikipedia. Located at : en.Wikipedia.org/wiki/Antigen_presentation . License : CC BY-SA: Attribution-ShareAlike
- cytotoxic. Provided by : Wiktionary. Located at : en.wiktionary.org/wiki/cytotoxic . License : CC BY-SA: Attribution-ShareAlike
- Antigen presentation. Provided by : Wikipedia. Located at : en.Wikipedia.org/wiki/Antigen_presentation . License : Public Domain: No Known Copyright

An official website of the United States government
The .gov means it's official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you're on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
- Browse Titles
NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Janeway CA Jr, Travers P, Walport M, et al. Immunobiology: The Immune System in Health and Disease. 5th edition. New York: Garland Science; 2001.
By agreement with the publisher, this book is accessible by the search feature, but cannot be browsed.

Immunobiology: The Immune System in Health and Disease. 5th edition.
Chapter 5 antigen presentation to t lymphocytes.
In an adaptive immune response , antigen is recognized by two distinct sets of highly variable receptor molecules—the immunoglobulins that serve as antigen receptors on B cells and the antigen-specific receptors of T cells . As we saw in Chapter 3, T cells recognize only antigens that are displayed on cell surfaces. These antigens may derive from pathogens that replicate within cells, such as viruses or intracellular bacteria , or from pathogens or their products that cells internalize by endocytosis from the extracellular fluid. T cells can detect the presence of intracellular pathogens because infected cells display on their surface peptide fragments derived from the pathogens' proteins. These foreign peptides are delivered to the cell surface by specialized host-cell glycoproteins, the MHC molecules , which are also described in Chapter 3. The MHC glycoproteins are encoded in a large cluster of genes that were first identified by their potent effects on the immune response to transplanted tissues. For that reason, the gene complex was termed the major histocompatibility complex (MHC) . We now know that within this region of the genome, in addition to those genes encoding the MHC molecules themselves, are many genes whose products are involved in the production of the MHC:peptide complexes.
We will begin by discussing the mechanisms of antigen processing and presentation, whereby protein antigens are degraded into peptides inside cells and the peptides are then carried to the cell surface bound to MHC molecules . We will see that the two different classes of MHC molecule, known as MHC class I and MHC class II, deliver peptides from different cellular compartments to the surface of the infected cell. Peptides from the cytosol are bound to MHC class I molecules and are recognized by CD8 T cells , whereas peptides generated in vesicles are bound to MHC class II molecules and recognized by CD4 T cells . The two functional subsets of T cells are thereby activated to initiate the destruction of pathogens resident in these two different cellular compartments. Some CD4 T cells activate naive B cells that have internalized specific antigen, and thus also stimulate the production of antibodies to extracellular pathogens and their products.
In the second part of this chapter we will see that there are several genes for each class of MHC molecule: that is, the MHC is polygenic . Each of these genes has many variants: that is, the MHC is also highly polymorphic. Indeed, the most remarkable feature of the MHC class I and II genes is their genetic variability . MHC polymorphism has a profound effect on antigen recognition by T cells , and the combination of polygeny and polymorphism greatly extends the range of peptides that can be presented to T cells by each individual and each population at risk from an infectious pathogen.
- The generation of T-cell receptor ligands
- The major histocompatibility complex and its functions
- Summary to Chapter 5
- General references
- Section references
- Cite this Page Janeway CA Jr, Travers P, Walport M, et al. Immunobiology: The Immune System in Health and Disease. 5th edition. New York: Garland Science; 2001. Chapter 5, Antigen Presentation to T Lymphocytes.
- Disable Glossary Links
Related Items in Bookshelf
- All Textbooks
Recent Activity
- Antigen Presentation to T Lymphocytes - Immunobiology Antigen Presentation to T Lymphocytes - Immunobiology
Your browsing activity is empty.
Activity recording is turned off.
Turn recording back on
Connect with NLM
National Library of Medicine 8600 Rockville Pike Bethesda, MD 20894
Web Policies FOIA HHS Vulnerability Disclosure
Help Accessibility Careers

- school Campus Bookshelves
- menu_book Bookshelves
- perm_media Learning Objects
- login Login
- how_to_reg Request Instructor Account
- hub Instructor Commons
- Download Page (PDF)
- Download Full Book (PDF)
- Periodic Table
- Physics Constants
- Scientific Calculator
- Reference & Cite
- Tools expand_more
- Readability
selected template will load here
This action is not available.
18.2: Antigens, Antigen Presenting Cells, and Major Histocompatibility Complexes
- Last updated
- Save as PDF
- Page ID 5222

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
Learning Objectives
- Identify cells that express MHC I and/or MHC II molecules and describe the structures and cellular location of MHC I and MHC II molecules
- Identify the cells that are antigen-presenting cells
- Describe the process of antigen processing and presentation with MHC I and MHC II
As discussed in Cellular Defenses , major histocompatibility complex (MHC) molecules are expressed on the surface of healthy cells, identifying them as normal and “self” to natural killer (NK) cells. MHC molecules also play an important role in the presentation of foreign antigens, which is a critical step in the activation of T cells and thus an important mechanism of the adaptive immune system.
Major Histocompatibility Complex Molecules
The major histocompatibility complex (MHC) is a collection of genes coding for MHC molecules found on the surface of all nucleated cells of the body. In humans, the MHC genes are also referred to as human leukocyte antigen (HLA) genes. Mature red blood cells, which lack a nucleus, are the only cells that do not express MHC molecules on their surface.
There are two classes of MHC molecules involved in adaptive immunity, MHC I and MHC II (Figure \(\PageIndex{1}\)). MHC I molecules are found on all nucleated cells; they present normal self-antigens as well as abnormal or nonself pathogens to the effector T cells involved in cellular immunity. In contrast, MHC II molecules are only found on macrophages, dendritic cells, and B cells; they present abnormal or nonself pathogen antigens for the initial activation of T cells.
Both types of MHC molecules are transmembrane glycoproteins that assemble as dimers in the cytoplasmic membrane of cells, but their structures are quite different. MHC I molecules are composed of a longer α protein chain coupled with a smaller β 2 microglobulin protein, and only the α chain spans the cytoplasmic membrane. The α chain of the MHC I molecule folds into three separate domains: α 1 , α 2 and α 3 . MHC II molecules are composed of two protein chains (an α and a β chain) that are approximately similar in length. Both chains of the MHC II molecule possess portions that span the plasma membrane, and each chain folds into two separate domains: α 1 and α 2 , and β 1 , and β 2 . In order to present abnormal or non-self-antigens to T cells, MHC molecules have a cleft that serves as the antigen-binding site near the “top” (or outermost) portion of the MHC-I or MHC-II dimer. For MHC I, the antigen-binding cleft is formed by the α 1 and α 2 domains, whereas for MHC II, the cleft is formed by the α 1 and β 1 domains (Figure \(\PageIndex{1}\)).

Exercise \(\PageIndex{1}\)
Compare the structures of the MHC I and MHC II molecules.
Antigen-Presenting Cells (APCs)
All nucleated cells in the body have mechanisms for processing and presenting antigens in association with MHC molecules. This signals the immune system, indicating whether the cell is normal and healthy or infected with an intracellular pathogen. However, only macrophages, dendritic cells, and B cells have the ability to present antigens specifically for the purpose of activating T cells; for this reason, these types of cells are sometimes referred to as antigen-presenting cells (APCs).
While all APCs play a similar role in adaptive immunity, there are some important differences to consider. Macrophages and dendritic cells are phagocytes that ingest and kill pathogens that penetrate the first-line barriers (i.e., skin and mucous membranes). B cells, on the other hand, do not function as phagocytes but play a primary role in the production and secretion of antibodies. In addition, whereas macrophages and dendritic cells recognize pathogens through nonspecific receptor interactions (e.g., PAMPs, toll-like receptors, and receptors for opsonizing complement or antibody), B cells interact with foreign pathogens or their free antigens using antigen-specific immunoglobulin as receptors (monomeric IgD and IgM). When the immunoglobulin receptors bind to an antigen, the B cell internalizes the antigen by endocytosis before processing and presentting the antigen to T cells.
Antigen Presentation with MHC II Molecules
MHC II molecules are only found on the surface of APCs. Macrophages and dendritic cells use similar mechanisms for processing and presentation of antigens and their epitopes in association with MHC II; B cells use somewhat different mechanisms that will be described further in B Lymphocytes and Humoral Immunity . For now, we will focus on the steps of the process as they pertain to dendritic cells.
After a dendritic cell recognizes and attaches to a pathogen cell, the pathogen is internalized by phagocytosis and is initially contained within a phagosome. Lysosomes containing antimicrobial enzymes and chemicals fuse with the phagosome to create a phagolysosome, where degradation of the pathogen for antigen processing begins. Proteases (protein-degrading) are especially important in antigen processing because only protein antigen epitopes are presented to T cells by MHC II (Figure \(\PageIndex{2}\)).
APCs do not present all possible epitopes to T cells; only a selection of the most antigenic or immunodominantepitopes are presented. The mechanism by which epitopes are selected for processing and presentation by an APC is complicated and not well understood; however, once the most antigenic, immunodominant epitopes have been processed, they associate within the antigen-binding cleft of MHC II molecules and are translocated to the cell surface of the dendritic cell for presentation to T cells.

Exercise \(\PageIndex{2}\)
- What are the three kinds of APCs?
- What role to MHC II molecules play in antigen presentation?
- What is the role of antigen presentation in adaptive immunity?
Antigen Presentation with MHC I Molecules
MHC I molecules, found on all normal, healthy, nucleated cells, signal to the immune system that the cell is a normal “self” cell. In a healthy cell, proteins normally found in the cytoplasm are degraded by proteasomes (enzyme complexes responsible for degradation and processing of proteins) and processed into self-antigen epitopes; these self-antigen epitopes bind within the MHC I antigen-binding cleft and are then presented on the cell surface. Immune cells, such as NK cells, recognize these self-antigens and do not target the cell for destruction. However, if a cell becomes infected with an intracellular pathogen (e.g., a virus), protein antigens specific to the pathogen are processed in the proteasomes and bind with MHC I molecules for presentation on the cell surface. This presentation of pathogen-specific antigens with MHC I signals that the infected cell must be targeted for destruction along with the pathogen.
Before elimination of infected cells can begin, APCs must first activate the T cells involved in cellular immunity. If an intracellular pathogen directly infects the cytoplasm of an APC, then the processing and presentation of antigens can occur as described (in proteasomes and on the cell surface with MHC I). However, if the intracellular pathogen does not directly infect APCs, an alternative strategy called cross-presentation is utilized. In cross-presentation, antigens are brought into the APC by mechanisms normally leading to presentation with MHC II (i.e., through phagocytosis), but the antigen is presented on an MHC I molecule for CD8 T cells. The exact mechanisms by which cross-presentation occur are not yet well understood, but it appears that cross-presentation is primarily a function of dendritic cells and not macrophages or B cells.
Exercise \(\PageIndex{3}\)
- Compare and contrast antigen processing and presentation associated with MHC I and MHC II molecules.
- What is cross-presentation, and when is it likely to occur?
Key Concepts and Summary
- Major histocompatibility complex (MHC) is a collection of genes coding for glycoprotein molecules expressed on the surface of all nucleated cells.
- MHC I molecules are expressed on all nucleated cells and are essential for presentation of normal “self” antigens. Cells that become infected by intracellular pathogens can present foreign antigens on MHC I as well, marking the infected cell for destruction.
- MHC II molecules are expressed only on the surface of antigen-presenting cells (macrophages, dendritic cells, and B cells). Antigen presentation with MHC II is essential for the activation of T cells.
- Antigen-presenting cells (APCs) primarily ingest pathogens by phagocytosis, destroy them in the phagolysosomes, process the protein antigens, and select the most antigenic/immunodominant epitopes with MHC II for presentation to T cells.
- Cross-presentation is a mechanism of antigen presentation and T-cell activation used by dendritic cells not directly infected by the pathogen; it involves phagocytosis of the pathogen but presentation on MHC I rather than MHC II.
Cancer immune escape: the role of antigen presentation machinery
- Open access
- Published: 09 April 2023
- Volume 149 , pages 8131–8141, ( 2023 )
Cite this article
You have full access to this open access article

- Anoop Kallingal ORCID: orcid.org/0000-0002-9613-3259 1 ,
- Mateusz Olszewski ORCID: orcid.org/0000-0002-1952-4985 1 ,
- Natalia Maciejewska ORCID: orcid.org/0000-0001-9942-285X 1 ,
- Wioletta Brankiewicz ORCID: orcid.org/0000-0002-8314-0775 1 , 2 &
- Maciej Baginski 1
5038 Accesses
10 Citations
Explore all metrics
The mechanisms of antigen processing and presentation play a crucial role in the recognition and targeting of cancer cells by the immune system. Cancer cells can evade the immune system by downregulating or losing the expression of the proteins recognized by the immune cells as antigens, creating an immunosuppressive microenvironment, and altering their ability to process and present antigens. This review focuses on the mechanisms of cancer immune evasion with a specific emphasis on the role of antigen presentation machinery. The study of the immunopeptidome, or peptidomics, has provided insights into the mechanisms of cancer immune evasion and has potential applications in cancer diagnosis and treatment. Additionally, manipulating the epigenetic landscape of cancer cells plays a critical role in suppressing the immune response against cancer. Targeting these mechanisms through the use of HDACis, DNMTis, and combination therapies has the potential to improve the efficacy of cancer immunotherapy. However, further research is needed to fully understand the mechanisms of action and optimal use of these therapies in the clinical setting.
Similar content being viewed by others

Cancer Immunoediting: Immunosurveillance, Immune Equilibrium, and Immune Escape

Primer on Cancer Immunotherapy and the Targeting of Native Proteins
Avoid common mistakes on your manuscript.
Introduction
The role of antigen presentation in cancer immune cell escape is a complex and multifaceted topic that has been the subject of much research in recent years. Antigen presentation is the process by which cells in the immune system display foreign molecules, such as those from pathogens or cancer cells, on their surface for recognition by other immune cells (Zitvogel and Kroemer 2018 ). In the context of cancer, antigen presentation plays a crucial role in the ability of the immune system to identify and target cancer cells. However, cancer cells can evade the immune system by various mechanisms, including downregulating or losing the expression of the proteins recognized by the immune cells as antigens, a process known as an immune escape (Beatty and Gladney 2015 ). The process of antigen presentation begins with the cancer cells expressing proteins on their surface, which are then recognized by specialized immune cells called antigen-presenting cells (APCs) (Mpakali and Stratikos 2021 ). These APCs, such as dendritic cells, then internalize the cancer cell proteins and degrade them into smaller peptides. These peptides are then displayed on the surface of the APC, along with particular proteins called major histocompatibility complex (MHC) molecules (Blum et al. 2013 ). The MHC molecules act as a bridge between the cancer cell proteins and the immune cells responsible for recognizing and attacking cancer cells, called T cells. The T cells have T cell receptors (TCRs) that can recognize the cancer cell proteins displayed on the MHC molecules (Alberts et al. 2002 ). When a T cell recognizes a cancer cell protein displayed on an APC, it becomes activated and begins to divide and differentiate into specialized cells that can attack and destroy the cancer cells (Messerschmidt et al. 2016 ). Cancer cells can evade the immune system by downregulating or losing the expression of the proteins recognized by the immune cells as antigens (Beatty and Gladney 2015 ). This can happen by mutations in the cancer cells that affect the expression of these proteins or by the cancer cells creating an immunosuppressive microenvironment that prevents the immune cells from recognizing and attacking the cancer cells (Brody 2016 ). Some cancer cells can produce molecules called immune checkpoint inhibitors that bind to and inhibit the activity of T cells, preventing them from recognizing and attacking cancer cells (Lao et al. 2022 ).
Additionally, cancer cells can recruit immune cells that promote immune suppression, such as regulatory T cells and myeloid-derived suppressor cells, which further dampen the immune response against cancer (Brody 2016 ). Cancer cells can also evade the immune system by changing the location of the antigens within the cell, called the abscopal effect, where the cancer cells move the antigens to the inside of the cell, making them invisible to the immune system (Beatty and Gladney 2015 ; Alfonso et al. 2020 ). Recent research has shown that targeting the mechanisms of antigen presentation and immune escape can be an effective strategy for treating cancer. For example, drugs that block immune checkpoint inhibitors, such as anti-CTLA-4 and anti-PD-1/PD-L1, have been approved for use in several types of cancer and have shown promising results in clinical trials (Seidel et al. 2018 ; Rotte 2019 ). In a snapshot, antigen presentation plays a crucial role in the ability of the immune system to identify and target cancer cells. Understanding the mechanisms of antigen presentation and immune escape is crucial for developing effective cancer immunotherapies.
Immune system and cancer
The immune system plays a crucial role in the development and progression of cancer (Gonzalez et al. 2018 ). Cancer cells develop from normal cells and can evade the immune system through various mechanisms; one of them is a process known as an immune escape. The immune system can recognize and target cancer cells through immunosurveillance. This process involves specialized immune cells, such as T cells and natural killer cells, that can detect and destroy cancer cells (Marcus et al. 2014 ; Gonzalez et al. 2018 ). The immune system also plays a role in shaping the microenvironment of the tumour. Tumour-associated macrophages, dendritic cells, and Treg cells are some of the cells found in the tumour microenvironment and play a role in cancer progression (Anderson and Simon 2020 ). Tumour-associated macrophages and dendritic cells can promote cancer cell growth by secreting factors that promote angiogenesis and inhibiting T cell activity. On the other hand Treg cells can suppress the immune response against cancer by inhibiting the activation and proliferation of T cells (Baay et al. 2011 ).
Another important mechanism in cancer progression is the ability of cancer cells to evade the immune system by downregulating or losing the expression of the proteins recognized by the immune cells as antigens (Dhatchinamoorthy et al. 2021 ). Recent research has shown that targeting the mechanisms of antigen presentation and immune escape can be an effective strategy for treating cancer. For example, drugs that block immune checkpoint inhibitors, such as anti-CTLA-4 and anti-PD-1/PD-L1, have been approved for use in several types of cancer and have shown promising results in clinical trials (Wojtukiewicz et al. 2021 ; Xiang et al. 2022 ; Sové et al. 2022 ). The immune system plays a crucial role in the development and progression of cancer. Understanding the mechanisms of immunosurveillance, immune escape, and the immune system's role in shaping the tumour microenvironment is crucial for developing effective cancer immunotherapies. Immune-based therapies, such as cancer vaccines and checkpoint inhibitors, have shown great promise in treating cancer and are expected to play a significant role in cancer treatment.
Immune checkpoints and immune evasion in cancer
Cancer immune evasion refers to the ability of cancer cells to evade detection and destruction by the immune system (Vinay et al. 2015 ). This complex process involves multiple mechanisms that enable cancer cells to evade the immunosurveillance mechanisms of the body (Messerschmidt et al. 2016 ).
Immune checkpoints are molecules or pathways that regulate the activation and function of the immune system. Immune checkpoint inhibitors are a class of drugs that block the function of these checkpoints, thereby enhancing the immune response against cancer cells (He and Xu 2020 ). One of the most well-known immune checkpoint pathways is the CTLA-4 pathway (Buchbinder and Desai 2016 ). CTLA-4 is a protein expressed on the surface of T cells that acts as an inhibitory receptor, blocking the activation and proliferation of T cells (Parry et al. 2005 ). Anti-CTLA-4 therapies, such as ipilimumab, act by binding to and blocking the function of CTLA-4, thereby enhancing the immune response against cancer cells (Callahan et al. 2010 ). Another critical immune checkpoint pathway is the PD-1/PD-L1. PD-1 is a protein expressed on the surface of T cells that interacts with PD-L1, which is expressed on the surface of cancer cells. This interaction blocks the activation and proliferation of T cells, allowing cancer cells to evade the immune response (Han et al. 2020 ). Anti-PD-1/PD-L1 treatments, such as nivolumab and pembrolizumab, work by binding to and inhibiting the interaction of PD-1 and PD-L1, increasing the immune response against cancer cells (Fessas et al. 2017 ) (Fig. 1 ).

Immune checkpoint inhibitors, such as anti-CTLA-4 and anti-PD-1/PD-L1 drugs, enhance the immune response against cancer by blocking immune checkpoint pathways. Other checkpoint pathways, such as LAG-3 and TIGIT, are being investigated as potential targets for cancer therapy and may have synergistic effects when combined with other checkpoint inhibitors
Other immune checkpoint pathways, such as LAG-3 and TIGIT, are also being investigated as potential targets for cancer therapy. LAG-3 (lymphocyte activation gene 3) is a protein that binds to MHC class II molecules and regulates T cell activation and exhaustion (Ge et al. 2021 ; Huo et al. 2022 ). TIGIT (T cell immunoreceptor with Ig and ITIM domains) is a protein that binds to both T cells and immune cells and regulates T cell activation and function. Preclinical research has demonstrated a significant impact of these pathways, and clinical trials are currently being conducted to explore their potential as therapeutic cancer targets (Yue et al. 2022 ). LAG-3 and TIGIT have a unique mechanism of action compared to other immune checkpoint inhibitors, such as PD-1 and CTLA-4, and may have a synergistic effect when combined with these drugs. This could potentially lead to improved efficacy and reduced side effects. In preclinical studies, TIGIT and LAG-3 inhibitors are effective in combination with PD-1 inhibitors in various cancer models, such as melanoma, lung cancer, and ovarian cancer (De Sousa et al. 2018 ; Seidel et al. 2018 ; Willsmore et al. 2021 ).
Antigen presentation in cancer
Antigen processing and presentation are crucial mechanisms by which the immune system recognizes and targets cancer cells. This process involves the recognition of cancer cell-associated antigens by APCs and their subsequent presentation on the surface of these cells in a form that can be recognized by T cells (Mpakali and Stratikos 2021 ). The antigen processing and presentation process begins with the internalization of cancer cell-associated antigens by APCs (Blum et al. 2013 ; Lee et al. 2020 ). Once inside the cell, the antigens are degraded into small peptides by a complex of enzymes called the proteasome. These peptides are then transported to the endoplasmic reticulum, complex with MHC molecules (Rock et al. 2010 ). MHC molecules are specialized proteins that are essential for the recognition of antigens by T cells. There are two main types of MHC molecules: MHC class I and MHC class II. MHC class I molecules are expressed on the surface of all nucleated cells, including cancer cells, and present peptides derived from intracellular antigens. On the other hand, MHC class II molecules are expressed primarily on the surface of APCs and present peptides derived from extracellular antigens (Wieczorek et al. 2017 ).
The MHC-peptide complex is then transported to the cell surface, where it can be recognized by T cells. T cells have specialized T cell receptors (TCRs) that recognize the MHC-peptide complex (Alberts et al. 2002 ). When a T cell recognizes a cancer cell-associated antigen displayed on an APC, it becomes activated and begins to divide and differentiate into specialized cells that can attack and destroy the cancer cells (Kunimasa and Goto 2020 ). However, cancer cells can evade the immune system by downregulating or losing the expression of the proteins recognized by the immune cells as antigens. This can happen by mutations in the cancer cells that affect the expression of these proteins or by the cancer cells creating an immunosuppressive microenvironment that prevents the immune cells from recognizing and attacking the cancer cells (Beatty and Gladney 2015 ) (Fig. 2 ). Many reports have shown that cancer cells can also evade the immune system by altering their ability to process and present antigens. For example, some cancer cells can downregulate the expression of MHC molecules, making them invisible to the immune system (Mittal et al. 2014 ; Reeves and James 2017 ; Kulkarni et al. 2019 ). Cancer cells can also interfere with the activity of the proteasome, thereby preventing the degradation of cancer cell-associated antigens (Mittal et al. 2014 ; Reeves and James 2017 ; Kulkarni et al. 2019 ).

APCs internalize cancer cell-associated antigens and degrade them into small peptides, which are then presented on the surface of APCs as MHC-peptide complexes that can be recognized by T cells. Cancer cells can evade the immune system by downregulating or losing the expression of antigen proteins, altering their ability to process and present antigens, or creating an immunosuppressive microenvironment
MHC 1 in antigen presentation
Major histocompatibility complex class I (MHC-I) molecules play a critical role in antigen presentation. These molecules are expressed on the surface of all nucleated cells, including cancer cells, and are responsible for the presentation of peptides derived from intracellular antigens to CD8 + T cells, also known as cytotoxic T cells (van den Elsen 2011 ; Wang et al. 2019 ). The MHC-I molecule comprises two main components: the heavy chain, encoded by the HLA gene, and the beta-2-microglobulin (β2m), a non-polymorphic component. The heavy chain comprises three main domains: the α1, α2, and α3. The α1 and α2 domains bind the MHC-I molecule to the peptide, while the α3 domain is responsible for interacting with the CD8 T-cell receptor (Cruz-Tapias et al. 2013 ). The process of MHC-I presentation begins with the internalization of antigens by the cell. Once an antigen enters a cell, a group of enzymes called the proteasome breaks it down into a little peptide.
Peptide loading delivers these peptides to the endoplasmic reticulum, where they interact with the MHC-I molecule. The MHC-I-peptide complex is then transported to the cell surface, where it can be recognized by CD8 + T cells (Hewitt 2003 ). The binding of the peptide to the MHC-I molecule is mediated by the peptide-binding groove, which is composed of the α1 and α2 domains. The peptide-binding groove can only bind to peptides that are 8–10 amino acids long. Once the peptide is bound to the MHC-I molecule, it is transported to the cell surface (Fig. 3 ) (Zacharias and Springer 2004 ). Downregulating or removing proteins that express antigens allows cancer cells to evade the immune system. The ability of cancer cells to process and present antigens on MHC-I molecules can change if they develop an immunosuppressive microenvironment or experience protein expression mutations. Understanding the mechanisms of MHC-I presentation in cancer is crucial for developing effective cancer immunotherapies.

MHC-I antigen presentation. MHC-I molecules on the cell surface present intracellular antigen peptides to CD8 + T cells. Cancer cells can evade the immune system by downregulating antigen expression or altering antigen processing and presentation on MHC-I
Immunopeptidome and cancer
The immunopeptidome is the set of peptides presented by MHC molecules on the surface of cells (Yewdell 2022a ). These peptides are derived from the degradation of intracellular proteins and are essential for recognizing cancer cells by the immune system. The study of the immunopeptidome, also known as peptidomics, has revealed insights into the mechanisms of cancer immune evasion and has potential applications in cancer diagnosis and treatment (Synowsky et al. 2017 ; Yewdell 2022b ). One of the critical roles of the immunopeptidome in cancer is its ability to identify unique peptides specific to cancer cells. These cancer-specific peptides, also known as neoantigens, can be used to develop personalized cancer vaccines targeting the unique mutations in an individual's cancer. Neoantigen-based vaccines have shown promising results in clinical trials and are expected to play an essential role in the future of cancer immunotherapy (D’Amico et al. 2022 ; Ouspenskaia et al. 2022 ). Another essential role of the immunopeptidome in cancer is its ability to provide insights into the mechanisms of cancer immune evasion. The study of the immunopeptidome can reveal which proteins are being presented by MHC molecules and which are not, providing insight into the mechanisms of cancer immune evasion (León-Letelier et al. 2022 ). The immunopeptidome can also provide valuable information for cancer diagnosis, such as immunopeptidome-based cancer diagnostics, tumour-associated antigen (TAA) testing, MHC class I tetramer staining and mass spectrometry-based peptidomics. Additionally, the study of the immunopeptidome can provide insights into the progression of cancer and the response to treatment by monitoring changes in the peptides presented by MHC molecules (Dersh et al. 2021 ).
Tumor antigen expression, presentation and control
The control of tumour antigen expression and presentation is a critical aspect of cancer biology that significantly impacts the immune system's ability to recognize and target cancer cells (Whiteside 2006 ). Tumours evade immune recognition through various mechanisms, such as the downregulation of antigens recognized by immune cells, the creation of an immunosuppressive microenvironment, and interaction with immune checkpoint pathways. Tumour antigens are molecules expressed on the surface of cancer cells and recognized by the immune system as foreign (Fig. 4 ).

Tumors can evade detection and destruction by the immune system, thereby allowing for uncontrolled growth and progression. This process is referred to as immune evasion and is a complex mechanism that involves the downregulation or loss of antigens recognized by immune cells, the creation of an immunosuppressive microenvironment, and interaction with immune checkpoint pathways
Cancer cells can regulate tumour antigen expression via epigenetics, like DNA structure changes (methylation, histone modification). They can also reduce antigen expression, hide from the immune system, and inhibit antigen-presenting cells/T cells (TGF-beta, IL-10) from suppressing immune response.(Gibney and Nolan 2010 ). Another mechanism by which cancer cells can control the expression of tumour antigens is through the manipulation of the proteasome and the MHC molecules (Boulpicante et al. 2020 ). The proteasome is a complex of enzymes responsible for degrading intracellular proteins, including tumour antigens, into peptides that MHC molecules can present. Cancer cells can interfere with the activity of the proteasome, thereby preventing the degradation of cancer cell-associated antigens and avoiding the presentation of the antigens on the MHC molecules (Chen et al. 2022 ). Cancer cells can also downregulate the expression of MHC molecules, thus making them invisible to the immune system and avoiding antigen presentation, or manipulate the structure of the MHC molecules, such as altering the peptide binding affinity, which can prevent the presentation of the cancer-associated antigens (Hewitt 2003 ; Rock et al. 2010 ; Blum et al. 2013 ).
Epigenetic modulation of immunotherapy
One mechanism by which cancer cells can control the expression of tumour antigens is through epigenetic regulation. Epigenetics refers to the regulation of gene expression through changes in the structure of DNA, such as methylation and histone modification, rather than changes in the genetic code itself (Gibney and Nolan 2010 ). Cancer cells can alter the epigenetic landscape to downregulate the expression of tumour antigens, making them invisible to the immune system. Cancer cells can also secrete factors that inhibit the activity of antigen-presenting cells and T cells, such as TGF-beta and IL-10, which further suppress the immune response (Thepmalee et al. 2018 ). Epigenetic modulation of antitumor immunity has been an active area of research in recent years and has been found to have potential applications in cancer immunotherapy (Gibney and Nolan 2010 ). Cancer cells' manipulation of the epigenetic landscape has been shown to play a critical role in suppressing the immune response against cancer. By targeting these mechanisms, it is possible to improve the efficacy of cancer immunotherapy (Liu et al. 2022a ). One way in which epigenetic modulation can be targeted is through the use of histone deacetylase inhibitors (HDACis). HDACis are a class of drugs that inhibit the activity of histone deacetylases, enzymes that remove acetyl groups from histones, leading to the repression of gene expression. HDACis have been shown to enhance the maturation of dendritic cells and increase the presentation of tumour antigens, thus enhancing the immune response against cancer (Gryder et al. 2012 ).
Another way to target epigenetic modulation is through DNA methyltransferase inhibitors (DNMTis) (Hu et al. 2021 ). DNMTis are a class of drugs that inhibit the activity of DNA methyltransferases, enzymes that add methyl groups to DNA, leading to the repression of gene expression. DNMTis have been shown to increase the expression of genes involved in the immune response, such as MHC molecules, and modulate the expression of genes involved in immune evasions, such as PD-L1 (Dan et al. 2019 ) (Fig. 5 ). The combination therapies that combine epigenetic modulation with other immunotherapeutic strategies, such as checkpoint inhibitors, have also yielded promising results in clinical trials. For example, combining HDACis with PD-1/PD-L1 inhibitors has enhanced the response to treatment in multiple cancer types (Mazzone et al. 2017 ; Liu et al. 2022b ).

Diagram illustrating the epigenetic regulation of chromatin accessibility and gene expression in cells. Nucleosomes, formed by DNA wrapped around histone octamers, are depicted as blue cylinders. Epigenetic modifications are depicted as dynamic interactions between chromatin components and enzymes, including histone methylation/demethylation, histone acetylation/deacetylation, and DNA methylation. Chromatin remodelling also plays a role in regulating gene expression
It is important to note that while the use of these epigenetic modulation therapies has shown promising results in preclinical and clinical studies, more research is needed to fully understand the mechanisms of action and optimal use in the clinical setting. Further research is also needed to understand these therapies' potential side effects and long-term safety.
Antigen presentation machinery components, modulation and their defects
The antigen processing machinery (APM) plays a critical role in developing an effective antitumor immune response (Maggs et al. 2021 ). The APM is a group of cellular structures and molecules responsible for processing and presenting APCs to T cells. Defects in the APM can compromise the ability of the immune system to recognize and respond to cancer cells, leading to the development of tumours that evade destruction by the immune system (Mpakali and Stratikos 2021 ). The major components of the APM include proteasomes, which are responsible for the degradation of proteins into peptides; TAP (transporter associated with antigen processing), which transports the peptides from the cytosol to the endoplasmic reticulum (ER); and MHC (major histocompatibility complex) molecules, which present the peptides on the surface of APCs to T cells. A growing body of evidence suggests that defects in the APM can contribute to cancer development. For example, mutations in the genes encoding the proteasomes or TAP can reduce the ability to generate peptides that can be presented on MHC molecules (Reiman et al. 2007 ). This can limit the ability of the immune system to recognize and respond to cancer cells. Additionally, defects in MHC molecules can result in a decreased ability to mount an immune response against certain infections and cancer (Charles et al. 2001 ; Dassa 2003 ).
Cancer cells can modulate antigen presentation in several ways to evade recognition and destruction by the immune system. Cancer cells can do this by deregulation of MHC molecules; Cancer cells can reduce the expression of MHC molecules on their surface, making them less visible to T cells and harder to target. Disruption of antigen processing; Cancer cells can interfere with the normal processing of antigens within the cell, making it harder for APCs to present them on MHC molecules. Production of immunosuppressive molecules; Cancer cells can produce molecules that suppress the immune response, such as TGF-beta and IDO, making it harder for T cells to recognize and attack cancer cells. Recruitment of immune-suppressive cells; Cancer cells can recruit immune cells that suppress the immune response, such as Tregs and MDSCs, to the tumour microenvironment (Vinay et al. 2015 ; Parcesepe et al. 2016 ; Mergener and Peña-Llopis 2022 ).
Defects in any of these components can result in a compromised immune response. For example, mutations in MHC molecules can result in a condition called MHC deficiency, which leads to a decreased ability to mount an immune response against certain infections. Similarly, TCR defects can result in T cell dysfunction and increased susceptibility to infections. Defects in the antigen presentation machinery can significantly impact the immune system's ability to recognize and respond to cancer cells, and understanding these defects can inform the development of new immunotherapies for cancer (Mpakali and Stratikos 2021 ). The development of immunotherapies for cancer has been a promising approach to targeting tumours that evade destruction by the immune system. These therapies aim to re-activate the patient's immune system to recognize and attack cancer cells. This can include checkpoint inhibitors, which block the immune-suppressive signals emitted by cancer cells and allow T cells to recognize and attack the tumour, and CAR T-cell therapy, which genetically modifies a patient's T cells to recognize and attack cancer cells (Filley et al. 2018 ).
Neoantigens in cancer immunotherapy
Neoantigens are a class of tumour-specific antigens generated by genetic mutations in cancer cells. They are not present in normal cells and, thus, represent a unique target for cancer immunotherapy. Identifying and characterising neoantigens have led to the development of new immunotherapeutic strategies for cancer treatment (Zhu and Liu 2021 ). The process of neoantigen identification begins with the sequencing of a patient's tumour and normal DNA (Zhu and Liu 2021 ). Algorithms are then used to identify potential neoantigens based on their predicted binding to MHC molecules and their potential to be presented on the cell surface. These potential neoantigens are further validated through functional assays, such as T-cell assays, to confirm their ability to elicit a T-cell response (Garcia-Garijo et al. 2019 ; Zaidi et al. 2020 ). Once identified, neoantigens can be used to develop personalized cancer vaccines (Blass and Ott 2021 ). These vaccines can target specific mutations in an individual's tumour and stimulate an immune response against cancer cells. The vaccines can be either ex vivo, where T cells are extracted from the patient, genetically modified to recognize the neoantigens, and then re-infused back into the patient or in vivo, where the patient is administered with the neoantigen peptides (Xie et al. 2023 ).
Recent clinical trials have demonstrated the safety and efficacy of personalized neoantigen cancer vaccines (Fritah et al. 2022 ). The results have shown that these vaccines can induce antitumor T-cell responses and result in durable clinical responses in a subset of patients with advanced cancer. Additionally, a combination of neoantigen vaccine with checkpoint inhibitors has shown to be more effective in inducing antitumor T-cell response and, in some cases, led to complete remission of the disease (Liao and Zhang 2021 ). Furthermore, the identification of neoantigens has also led to the development of neoantigen-targeting T-cell therapies, such as CAR-T cell therapy. In this approach, T cells are genetically modified to express a CAR specific for a neoantigen and then re-infused back into the patient. These therapies have shown effective in inducing long-lasting responses in patients with advanced cancer (Wang and Cao 2020 ).
The antigen processing and presentation mechanisms play a critical role in the immune system's recognition and targeting of cancer cells. Cancer cells can avoid immune detection by downregulating or losing the expression of proteins recognised as antigens, creating an immunosuppressive microenvironment, and altering their ability to process and present antigens. The study of the immunopeptidome, or peptidomics, has provided insights into the mechanisms of cancer immune evasion and has potential applications in cancer diagnosis and treatment. One mechanism by which cancer cells can control the expression of tumour antigens is through epigenetic regulation, such as methylation and histone modification; cancer cells can alter the epigenetic landscape to downregulate the expression of tumour antigens, making them invisible to the immune system. Additionally, cancer cells can manipulate the microenvironment, interfere with the activity of the proteasome and MHC molecules, and downregulate the expression of MHC molecules to avoid the presentation of antigens. Recent advances in cancer genomics and molecular biology have allowed the identification of unique antigens present in cancer cells but not in normal cells, known as "neoantigens." These neoantigens can be used to develop cancer vaccines and CAR-T cell therapy that target the specific mutations present in an individual's tumour, leading to the re-activation of the patient's immune system to recognize and attack cancer cells. Targeting the epigenetic mechanisms that cancer cells use to evade the immune system can improve cancer immunotherapy, such as using HDACis, DNMTis, and combination therapies. However, it's important to note that more research is needed to fully understand the mechanisms of action and optimal use of these therapies in the clinical setting. In snapshot, controlling tumour antigen expression and presentation is a critical aspect of cancer biology that significantly impacts the immune system's ability to recognize and target cancer cells. Understanding these mechanisms is crucial for developing effective cancer immunotherapies that target the mechanisms of antigen expression and presentation in cancer cells and for a better understanding of the epigenetic modulation of antitumor immunity for improved cancer immunotherapy.
Data availability
All data generated or analysed during this study are included in this published article.
Alberts B, Johnson A, Lewis J et al (2002) T cells and MHC proteins. Molecular biology of the cell, 4th edn. Garland Science, New York
Google Scholar
Alfonso JCL, Papaxenopoulou LA, Mascheroni P et al (2020) On the immunological consequences of conventionally fractionated radiotherapy. iScience 23:100897. https://doi.org/10.1016/j.isci.2020.100897
Anderson NM, Simon MC (2020) Tumor microenvironment. Curr Biol 30:R921–R925. https://doi.org/10.1016/j.cub.2020.06.081
Article CAS PubMed PubMed Central Google Scholar
Baay M, Brouwer A, Pauwels P et al (2011) Tumor cells and tumor-associated macrophages: secreted proteins as potential targets for therapy. Clin Dev Immunol 2011:565187. https://doi.org/10.1155/2011/565187
Beatty GL, Gladney WL (2015) Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res 21:687–692. https://doi.org/10.1158/1078-0432.CCR-14-1860
Article CAS PubMed Google Scholar
Blass E, Ott PA (2021) Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat Rev Clin Oncol 18:215–229. https://doi.org/10.1038/s41571-020-00460-2
Article PubMed PubMed Central Google Scholar
Blum JS, Wearsch PA, Cresswell P (2013) Pathways of antigen processing. Annu Rev Immunol 31:443–473. https://doi.org/10.1146/annurev-immunol-032712-095910
Boulpicante M, Darrigrand R, Pierson A et al (2020) Tumors escape immunosurveillance by overexpressing the proteasome activator PSME3. Oncoimmunology 9:1761205. https://doi.org/10.1080/2162402X.2020.1761205
Brody T (2016) Chapter 27-Mechanism of action—part II (cancer). In: Brody T (ed) Clinical trials, 2nd edn. Academic Press, Boston, pp 595–609
Chapter Google Scholar
Buchbinder EI, Desai A (2016) CTLA-4 and PD-1 pathways. Am J Clin Oncol 39:98–106. https://doi.org/10.1097/COC.0000000000000239
Callahan MK, Wolchok JD, Allison JP (2010) Anti-CTLA-4 antibody therapy: immune monitoring during clinical development of a novel immunotherapy. Semin Oncol 37:473–484. https://doi.org/10.1053/j.seminoncol.2010.09.001
Charles A Janeway J, Travers P, Walport M, Shlomchik MJ (2001) The major histocompatibility complex and its functions. Immunobiol Immune Syst Health Dis 5th Ed 1:1
Chen B, Zhu H, Yang B, Cao J (2022) The dichotomous role of immunoproteasome in cancer: Friend or foe? Acta Pharmaceut Sin B. https://doi.org/10.1016/j.apsb.2022.11.005
Article Google Scholar
Cruz-Tapias P, Castiblanco J, Anaya J-M (2013) Major histocompatibility complex: antigen processing and presentation. El Rosario University Press, London
D’Amico S, Tempora P, Melaiu O et al (2022) Targeting the antigen processing and presentation pathway to overcome resistance to immune checkpoint therapy. Front Immunol 13:1
Dan H, Zhang S, Zhou Y, Guan Q (2019) DNA methyltransferase inhibitors: catalysts for antitumour immune responses. Onco Targets Ther 12:10903–10916. https://doi.org/10.2147/OTT.S217767
Dassa E (2003) Chapter 1-Phylogenetic and functional classification of abc (atp-binding cassette) systems**abscisse, a database of ABC systems, which includes functional, sequence and structural information, is available on the internet at the following address: www.pasteur.fr/recherche/unites/pmtg/abc/index.html. In: Holland IB, Cole SPC, Kuchler K, Higgins CF (eds) ABC proteins. Academic Press, London, pp 3–35
De Sousa LA, Leitner J, Grabmeier-Pfistershammer K, Steinberger P (2018) Not all immune checkpoints are created equal. Front Immunol 9:1
Dersh D, Hollý J, Yewdell JW (2021) A few good peptides: MHC class I-based cancer immunosurveillance and immunoevasion. Nat Rev Immunol 21:116–128. https://doi.org/10.1038/s41577-020-0390-6
Dhatchinamoorthy K, Colbert JD, Rock KL (2021) Cancer immune evasion through loss of MHC class I antigen presentation. Front Immunol 12:636568. https://doi.org/10.3389/fimmu.2021.636568
Fessas P, Lee H, Ikemizu S, Janowitz T (2017) A molecular and preclinical comparison of the PD-1–targeted T-cell checkpoint inhibitors nivolumab and pembrolizumab. Semin Oncol 44:136–140. https://doi.org/10.1053/j.seminoncol.2017.06.002
Filley AC, Henriquez M, Dey M (2018) CART immunotherapy: development, success, and translation to malignant gliomas and other solid tumors. Front Oncol 8:453. https://doi.org/10.3389/fonc.2018.00453
Fritah H, Rovelli R, Chiang CL-L, Kandalaft LE (2022) The current clinical landscape of personalized cancer vaccines. Cancer Treat Rev. https://doi.org/10.1016/j.ctrv.2022.102383
Garcia-Garijo A, Fajardo CA, Gros A (2019) Determinants for neoantigen identification. Front Immunol 10:1
Ge Z, Peppelenbosch MP, Sprengers D, Kwekkeboom J (2021) TIGIT, the next step towards successful combination immune checkpoint therapy in cancer. Front Immunol 12:1
Gibney ER, Nolan CM (2010) Epigenetics and gene expression. Heredity 105:4–13. https://doi.org/10.1038/hdy.2010.54
Gonzalez H, Hagerling C, Werb Z (2018) Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev 32:1267. https://doi.org/10.1101/gad.314617.118
Gryder BE, Sodji QH, Oyelere AK (2012) Targeted cancer therapy: giving histone deacetylase inhibitors all they need to succeed. Future Med Chem 4:505–524. https://doi.org/10.4155/fmc.12.3
Han Y, Liu D, Li L (2020) PD-1/PD-L1 pathway: current researches in cancer. Am J Cancer Res 10:727–742
CAS PubMed PubMed Central Google Scholar
He X, Xu C (2020) Immune checkpoint signaling and cancer immunotherapy. Cell Res 30:660–669. https://doi.org/10.1038/s41422-020-0343-4
Hewitt EW (2003) The MHC class I antigen presentation pathway: strategies for viral immune evasion. Immunology 110:163–169. https://doi.org/10.1046/j.1365-2567.2003.01738.x
Hu C, Liu X, Zeng Y et al (2021) DNA methyltransferase inhibitors combination therapy for the treatment of solid tumor: mechanism and clinical application. Clin Epigenetics 13:166. https://doi.org/10.1186/s13148-021-01154-x
Huo J-L, Wang Y-T, Fu W-J et al (2022) The promising immune checkpoint LAG-3 in cancer immunotherapy: from basic research to clinical application. Front Immunol 13:1
Kulkarni B, Kirave P, Gondaliya P et al (2019) Exosomal miRNA in chemoresistance, immune evasion, metastasis and progression of cancer. Drug Discovery Today 24:2058–2067. https://doi.org/10.1016/j.drudis.2019.06.010
Kunimasa K, Goto T (2020) Immunosurveillance and immunoediting of lung cancer: current perspectives and challenges. Int J Mol Sci 21:597. https://doi.org/10.3390/ijms21020597
Lao Y, Shen D, Zhang W et al (2022) Immune checkpoint inhibitors in cancer therapy—How to overcome drug resistance? Cancers (basel) 14:3575. https://doi.org/10.3390/cancers14153575
Lee MY, Jeon JW, Sievers C, Allen CT (2020) Antigen processing and presentation in cancer immunotherapy. J Immunother Cancer 8:e001111. https://doi.org/10.1136/jitc-2020-001111
León-Letelier RA, Katayama H, Hanash S (2022) Mining the immunopeptidome for antigenic peptides in cancer. Cancers 14:4968. https://doi.org/10.3390/cancers14204968
Liao J-Y, Zhang S (2021) Safety and efficacy of personalized cancer vaccines in combination with immune checkpoint inhibitors in cancer treatment. Front Oncol 11:663264. https://doi.org/10.3389/fonc.2021.663264
Liu Z, Ren Y, Weng S et al (2022b) A new trend in cancer treatment: the combination of epigenetics and immunotherapy. Front Immunol 13:1
Liu Z, Ren Y, Weng S et al (2022a) A new trend in cancer treatment: the combination of epigenetics and immunotherapy. Front Immunol 13:809761. https://doi.org/10.3389/fimmu.2022.809761
Maggs L, Sadagopan A, Moghaddam AS, Ferrone S (2021) HLA class I antigen processing machinery defects in antitumor immunity and immunotherapy. Trends Cancer 7:1089–1101. https://doi.org/10.1016/j.trecan.2021.07.006
Marcus A, Gowen BG, Thompson TW et al (2014) Recognition of tumors by the innate immune system and natural killer cells. Adv Immunol 122:91–128. https://doi.org/10.1016/B978-0-12-800267-4.00003-1
Mazzone R, Zwergel C, Mai A, Valente S (2017) Epi-drugs in combination with immunotherapy: a new avenue to improve anticancer efficacy. Clin Epigenetics 9:59. https://doi.org/10.1186/s13148-017-0358-y
Mergener S, Peña-Llopis S (2022) A new perspective on immune evasion: escaping immune surveillance by inactivating tumor suppressors. Sig Transduct Target Ther 7:1–2. https://doi.org/10.1038/s41392-022-00875-6
Messerschmidt JL, Prendergast GC, Messerschmidt GL (2016) How cancers escape immune destruction and mechanisms of action for the new significantly active immune therapies: helping nonimmunologists decipher recent advances. Oncologist 21:233–243. https://doi.org/10.1634/theoncologist.2015-0282
Mittal D, Gubin MM, Schreiber RD, Smyth MJ (2014) New insights into cancer immunoediting and its three component phases—elimination, equilibrium and escape. Curr Opin Immunol 27:16–25. https://doi.org/10.1016/j.coi.2014.01.004
Mpakali A, Stratikos E (2021) The role of antigen processing and presentation in cancer and the efficacy of immune checkpoint inhibitor immunotherapy. Cancers (basel) 13:134. https://doi.org/10.3390/cancers13010134
Ouspenskaia T, Law T, Clauser KR et al (2022) Unannotated proteins expand the MHC-I-restricted immunopeptidome in cancer. Nat Biotechnol 40:209–217. https://doi.org/10.1038/s41587-021-01021-3
Parcesepe P, Giordano G, Laudanna C et al (2016) Cancer-associated immune resistance and evasion of immune surveillance in colorectal cancer. Gastroenterol Res Pract 2016:6261721. https://doi.org/10.1155/2016/6261721
Parry RV, Chemnitz JM, Frauwirth KA et al (2005) CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol 25:9543–9553. https://doi.org/10.1128/MCB.25.21.9543-9553.2005
Reeves E, James E (2017) Antigen processing and immune regulation in the response to tumours. Immunology 150:16–24. https://doi.org/10.1111/imm.12675
Reiman JM, Kmieciak M, Manjili MH, Knutson KL (2007) Tumor immunoediting and immunosculpting pathways to cancer progression. Semin Cancer Biol 17:275–287. https://doi.org/10.1016/j.semcancer.2007.06.009
Rock KL, Farfán-Arribas DJ, Shen L (2010) Proteases in MHC class I presentation and cross-presentation. J Immunol 184:9–15. https://doi.org/10.4049/jimmunol.0903399
Rotte A (2019) Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J Exp Clin Cancer Res 38:255. https://doi.org/10.1186/s13046-019-1259-z
Seidel JA, Otsuka A, Kabashima K (2018) Anti-PD-1 and Anti-CTLA-4 therapies in cancer: mechanisms of action, efficacy, and limitations. Front Oncol 8:86. https://doi.org/10.3389/fonc.2018.00086
Sové RJ, Verma BK, Wang H et al (2022) Virtual clinical trials of anti-PD-1 and anti-CTLA-4 immunotherapy in advanced hepatocellular carcinoma using a quantitative systems pharmacology model. J Immunother Cancer 10:e005414. https://doi.org/10.1136/jitc-2022-005414
Synowsky SA, Shirran SL, Cooke FGM et al (2017) The major histocompatibility complex class I immunopeptidome of extracellular vesicles. J Biol Chem 292:17084–17092. https://doi.org/10.1074/jbc.M117.805895
Thepmalee C, Panya A, Junking M et al (2018) Inhibition of IL-10 and TGF-β receptors on dendritic cells enhances activation of effector T-cells to kill cholangiocarcinoma cells. Hum Vaccin Immunother 14:1423–1431. https://doi.org/10.1080/21645515.2018.1431598
van den Elsen P (2011) Expression regulation of major histocompatibility complex class I and class II encoding genes. Front Immunol 2:1
Vinay DS, Ryan EP, Pawelec G et al (2015) Immune evasion in cancer: mechanistic basis and therapeutic strategies. Semin Cancer Biol 35:S185–S198. https://doi.org/10.1016/j.semcancer.2015.03.004
Wang Z, Cao YJ (2020) Adoptive cell therapy targeting neoantigens: a frontier for cancer research. Front Immunol 11:1
Wang Y, Tian M, Wang F et al (2019) Understanding the immunological mechanisms of mesenchymal stem cells in allogeneic transplantation: from the aspect of major histocompatibility complex class I. Stem Cells Dev 28:1141–1150. https://doi.org/10.1089/scd.2018.0256
Article PubMed Google Scholar
Whiteside TL (2006) Immune suppression in cancer: effects on immune cells, mechanisms and future therapeutic intervention. Semin Cancer Biol 16:3–15. https://doi.org/10.1016/j.semcancer.2005.07.008
Wieczorek M, Abualrous ET, Sticht J et al (2017) Major histocompatibility complex (MHC) class I and MHC class II proteins: conformational plasticity in antigen presentation. Front Immunol 8:1
Willsmore ZN, Coumbe BGT, Crescioli S et al (2021) Combined anti-PD-1 and anti-CTLA-4 checkpoint blockade: treatment of melanoma and immune mechanisms of action. Eur J Immunol 51:544–556. https://doi.org/10.1002/eji.202048747
Wojtukiewicz MZ, Rek MM, Karpowicz K et al (2021) Inhibitors of immune checkpoints—PD-1, PD-L1, CTLA-4—new opportunities for cancer patients and a new challenge for internists and general practitioners. Cancer Metastasis Rev 40:949–982. https://doi.org/10.1007/s10555-021-09976-0
Xiang Z, Li J, Zhang Z et al (2022) Comprehensive evaluation of anti-PD-1, Anti-PD-L1, Anti-CTLA-4 and their combined immunotherapy in clinical trials: a systematic review and meta-analysis. Front Pharmacol 13:1
Xie N, Shen G, Gao W et al (2023) Neoantigens: promising targets for cancer therapy. Sig Transduct Target Ther 8:1–38. https://doi.org/10.1038/s41392-022-01270-x
Article CAS Google Scholar
Yewdell JW (2022a) MHC class i immunopeptidome: past, present, and future. Mol Cell Proteom 21:100230. https://doi.org/10.1016/j.mcpro.2022.100230
Yewdell JW (2022b) MHC class i immunopeptidome: past, present, and future. Mol Cell Proteom 21:100230. https://doi.org/10.1016/j.mcpro.2022.100230
Yue C, Gao S, Li S et al (2022) TIGIT as a promising therapeutic target in autoimmune diseases. Front Immunol 13:1
Zacharias M, Springer S (2004) Conformational flexibility of the MHC class I α1-α2 domain in peptide bound and free states: a molecular dynamics simulation study. Biophys J 87:2203–2214. https://doi.org/10.1529/biophysj.104.044743
Zaidi N, Soban M, Chen F et al (2020) Role of in silico structural modeling in predicting immunogenic neoepitopes for cancer vaccine development. JCI Insight 5:e136991. https://doi.org/10.1172/jci.insight.136991
Zhu Y, Liu J (2021) The role of neoantigens in cancer immunotherapy. Front Oncol 11:682325. https://doi.org/10.3389/fonc.2021.682325
Zitvogel L, Kroemer G (eds) (2018). Springer, Cham
Download references
Acknowledgements
Conceptualization, methodology, writing original draft preparation, AK, MO, NM. writing—review and editing, WB. supervision, MB. All authors have read and agreed to the final version of the manuscript.
No funding was received to assist with the preparation of this manuscript.
Author information
Authors and affiliations.
Department of Pharmaceutical Technology and Biochemistry, Faculty of Chemistry, Gdansk University of Technology, Narutowicza St 11/12, 80-233, Gdansk, Poland
Anoop Kallingal, Mateusz Olszewski, Natalia Maciejewska, Wioletta Brankiewicz & Maciej Baginski
Department of Medical Genetics, Institute of Clinical Medicine, University of Oslo, Oslo, Norway
Wioletta Brankiewicz
You can also search for this author in PubMed Google Scholar
Contributions
The authors confirms sole responsibility for the following: study conception and design, data collection, analysis and interpretation of results, and manuscript preparation.
Corresponding author
Correspondence to Anoop Kallingal .
Ethics declarations
Conflict of interest.
The authors declare that they have no conflict of interest.
Additional information
Publisher's note.
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/ .
Reprints and permissions
About this article
Kallingal, A., Olszewski, M., Maciejewska, N. et al. Cancer immune escape: the role of antigen presentation machinery. J Cancer Res Clin Oncol 149 , 8131–8141 (2023). https://doi.org/10.1007/s00432-023-04737-8
Download citation
Received : 08 February 2023
Accepted : 31 March 2023
Published : 09 April 2023
Issue Date : August 2023
DOI : https://doi.org/10.1007/s00432-023-04737-8
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- Epigenetic modulation
- Immunopeptidome
- Neoantigens
- Find a journal
- Publish with us
- Track your research

You have no alerts at this time
- Product Catalog
- Member Directory

Patient Care and Research
- Guidelines and Consensus Documents
- Patient Safety
- Funding Opportunities
- Scientific Challenges
- Professional Development
- Provider Resources
Clinical Benefit From Immunotherapy in Patients With SCLC Is Associated With Tumor Capacity for Antigen Presentation
Affiliations.
- 1 Thoracic Oncology Service, Department of Medicine, Memorial Sloan Kettering Cancer Center, New York, New York; Department of Medicine, Weill Cornell Medical College, New York, New York; Druckenmiller Center for Lung Cancer Research, Memorial Sloan Kettering Cancer Center, New York, New York. Electronic address: [email protected].
- 2 Bristol-Myers Squibb, New York, New York.
- 3 Thoracic Oncology Service, Department of Medicine, Memorial Sloan Kettering Cancer Center, New York, New York; Department of Medicine, Weill Cornell Medical College, New York, New York.
- 4 Marie-Josée and Henry R. Kravis Center for Molecular Oncology, Memorial Sloan Kettering Cancer Center, New York, New York.
- 5 Cancer Biology Program, Louis V. Gerstner Jr. Graduate School of Biomedical Sciences, Memorial Sloan Kettering Cancer Center, New York, New York.
- 6 Druckenmiller Center for Lung Cancer Research, Memorial Sloan Kettering Cancer Center, New York, New York.
- 7 Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York.
- 8 Perlmutter Cancer Center, New York University Langone Health, New York, New York.
- PMID: 37210008
- PMCID: PMC10524620 (available on 2024-09-01 )
- DOI: 10.1016/j.jtho.2023.05.008
Introduction: A small percentage of patients with SCLC experience durable responses to immune checkpoint blockade (ICB). Defining determinants of immune response may nominate strategies to broaden the efficacy of immunotherapy in patients with SCLC. Prior studies have been limited by small numbers or concomitant chemotherapy administration.
Methods: CheckMate 032, a multicenter, open-label, phase 1/2 trial evaluating nivolumab alone or with ipilimumab was the largest study of ICB alone in patients with SCLC. We performed comprehensive RNA sequencing of 286 pretreatment SCLC tumor samples, assessing outcome on the basis of defined SCLC subtypes (SCLC-A, -N, -P, and -Y), and expression signatures associated with durable benefit, defined as progression-free survival more than or equal to 6 months. Potential biomarkers were further explored by immunohistochemistry.
Results: None of the subtypes were associated with survival. Antigen presentation machinery signature (p = 0.000032) and presence of more than or equal to 1% infiltrating CD8+ T cells by immunohistochemistry (hazard ratio = 0.51, 95% confidence interval: 0.27-0.95) both correlated with survival in patients treated with nivolumab. Pathway enrichment analysis revealed the association between durable benefit from immunotherapy and antigen processing and presentation. Analysis of epigenetic determinants of antigen presentation identified LSD1 gene expression as a correlate of worse survival outcomes for patients treated with either nivolumab or the combination of nivolumab and ipilimumab.
Conclusions: Tumor antigen processing and presentation is a key correlate of ICB efficacy in patients with SCLC. As antigen presentation machinery is frequently epigenetically suppressed in SCLC, this study defines a targetable mechanism by which we might improve clinical benefit of ICB for patients with SCLC.
Keywords: Antigen presentation; Epigenetics; Immune checkpoint blockade; Small cell lung cancer.
Copyright © 2023 International Association for the Study of Lung Cancer. Published by Elsevier Inc. All rights reserved.
Publication types
- Clinical Trial, Phase II
- Clinical Trial, Phase I
- Multicenter Study
- Research Support, Non-U.S. Gov't
- Research Support, N.I.H., Extramural
- Antigen Presentation
- Immunotherapy
- Ipilimumab / therapeutic use
- Lung Neoplasms* / pathology
- Nivolumab / therapeutic use
- Small Cell Lung Carcinoma* / pathology
Grants and funding
- P30 CA008748/CA/NCI NIH HHS/United States
- R01 CA258784/CA/NCI NIH HHS/United States
- R35 CA263816/CA/NCI NIH HHS/United States
- U24 CA213274/CA/NCI NIH HHS/United States
Log in using your username and password
- Search More Search for this keyword Advanced search
- Latest content
- Current issue
- BMJ Journals More You are viewing from: Google Indexer
You are here
- Online First
- Identification and validation of anti-protein arginine methyltransferase 5 (PRMT5) antibody as a novel biomarker for systemic sclerosis (SSc)
- Article Text
- Article info
- Citation Tools
- Rapid Responses
- Article metrics
- http://orcid.org/0000-0002-7650-1237 Minrui Liang 1 , 2 , 3 ,
- Lingbiao Wang 1 , 2 , 3 ,
- Xiaolong Tian 4 ,
- Kun Wang 5 , 6 ,
- Xiaoyi Zhu 4 ,
- Linlin Huang 1 , 2 , 3 ,
- Qing Li 5 , 6 ,
- Wenjing Ye 1 , 2 , 3 ,
- Chen Chen 1 , 2 , 7 ,
- Haihua Yang 8 ,
- Wanqing Wu 9 ,
- Xiangjun Chen 9 ,
- Xiaoxia Zhu 1 , 2 , 3 ,
- Yu Xue 1 , 2 , 3 ,
- Weiguo Wan 1 , 2 , 3 ,
- Yanling Wu 4 ,
- http://orcid.org/0000-0002-8634-0967 Liwei Lu 10 ,
- http://orcid.org/0000-0003-2765-0620 Jiucun Wang 11 ,
- Hejian Zou 1 , 2 , 3 ,
- Tianlei Ying 4 ,
- Feng Zhou 5 , 6
- 1 Department of Rheumatology , Huashan Hospital, Fudan University , Shanghai , China
- 2 Institute of Rheumatology, Immunology and Allergy , Fudan University , Shanghai , China
- 3 Huashan Rare Disease Center , Huashan Hospital, Fudan University , Shanghai , China
- 4 Key Laboratory of Medical Molecular Virology (MOE/NHC/CAMS) and Shanghai Institute of Infectious Disease and Biosecurity, Shanghai Frontiers Science Center of Pathogenic Microorganisms and Infection, Shanghai Engineering Research Center for Synthetic Immunology, School of Basic Medical Sciences , Fudan University , Shanghai , China
- 5 Shanghai Key Laboratory of Medical Epigenetics, International Co-laboratory of Medical Epigenetics and Metabolism, Ministry of Science and Technology, Institutes of Biomedical Sciences , Fudan University , Shanghai , China
- 6 Key Laboratory of Carcinogenesis and Cancer Invasion, Ministry of Education, Liver Cancer Institute, Zhongshan Hospital , Fudan University , Shanghai , China
- 7 Department of Emergency Medicine , Zhongshan Hospital, Fudan University , Shanghai , China
- 8 Department of Respiratory and Critical Care Medicine , Huashan Hospital, Fudan University , Shanghai , China
- 9 Department of Neurology , Huashan Hospital, Fudan University , Shanghai , China
- 10 Department of Pathology , The University of Hong Kong , Hong Kong , China
- 11 State Key Laboratory of Genetic Engineering, Collaborative Innovation Center for Genetics and Development, School of Life Sciences, and Human Phenome Institute , Fudan University , Shanghai , China
- Correspondence to Dr Minrui Liang, Department of Rheumatology, Institute of Rheumatology, Immunology and Allergy; Huashan Rare Disease Center, Huashan Hospital, Fudan University, Shanghai, China; mliang10{at}fudan.edu.cn ; Professor Tianlei Ying, Key Laboratory of Medical Molecular Virology (MOE/NHC/CAMS) and Shanghai Institute of Infectious Disease and Biosecurity, Shanghai Frontiers Science Center of Pathogenic Microorganisms and Infection, Shanghai Engineering Research Center for Synthetic Immunology, School of Basic Medical Sciences, Fudan University, Shanghai, China; tlying{at}fudan.edu.cn ; Professor Feng Zhou, Shanghai Key Laboratory of Medical Epigenetics, International Co-laboratory of Medical Epigenetics and Metabolism, Ministry of Science and Technology, Institutes of Biomedical Sciences, Fudan University, and Key Laboratory of Carcinogenesis and Cancer Invasion, Ministry of Education, Liver Cancer Institute, Zhongshan Hospital, Fudan University, Shanghai, China; zhou_feng{at}fudan.edu.cn
Objectives In the complex panorama of autoimmune diseases, the characterisation of pivotal contributing autoantibodies that are involved in disease progression remains challenging. This study aimed to employ a global antibody profiling strategy to identify novel antibodies and investigate their association with systemic sclerosis (SSc).
Methods We implemented this strategy by conducting immunoprecipitation (IP) following on-bead digestion with the sera of patients with SSc or healthy donors, using antigen pools derived from cell lysates. The enriched antigen-antibody complex was proceeded with mass spectrometry (MS)-based quantitative proteomics and over-represented by bioinformatics analysis. The candidate antibodies were then orthogonally validated in two independent groups of patients with SSc. Mice were immunised with the target antigen, which was subsequently evaluated by histological examination and RNA sequencing.
Results The IP-MS analysis, followed by validation in patients with SSc, revealed a significant elevation in anti-PRMT5 antibodies among patients with SSc. These antibodies exhibited robust diagnostic accuracy in distinguishing SSc from healthy controls and other autoimmune conditions, including systemic lupus erythematosus and Sjögren’s syndrome, with an area under the curve ranging from 0.900 to 0.988. The elevation of anti-PRMT5 antibodies was verified in a subsequent independent group with SSc using an additional method, microarray. Notably, 31.11% of patients with SSc exhibited seropositivity for anti-PRMT5 antibodies. Furthermore, the titres of anti-PRMT5 antibodies demonstrated a correlation with the progression or regression trajectory in SSc. PRMT5 immunisation displayed significant inflammation and fibrosis in both the skin and lungs of mice. This was concomitant with the upregulation of multiple proinflammatory and profibrotic pathways, thereby underscoring a potentially pivotal role of anti-PRMT5 antibodies in SSc.
Conclusions This study has identified anti-PRMT5 antibodies as a novel biomarker for SSc.
- Autoantibodies
- Autoimmune Diseases
- Scleroderma, Systemic
Data availability statement
Data are available upon reasonable request. All data relevant to the study are presented in the article or uploaded as supplementary information.
This is an open access article distributed in accordance with the Creative Commons Attribution 4.0 Unported (CC BY 4.0) license, which permits others to copy, redistribute, remix, transform and build upon this work for any purpose, provided the original work is properly cited, a link to the licence is given, and indication of whether changes were made. See: https://creativecommons.org/licenses/by/4.0/ .
https://doi.org/10.1136/ard-2024-225596
Statistics from Altmetric.com
Request permissions.
If you wish to reuse any or all of this article please use the link below which will take you to the Copyright Clearance Center’s RightsLink service. You will be able to get a quick price and instant permission to reuse the content in many different ways.
WHAT IS ALREADY KNOWN ON THIS TOPIC
The involvement of autoantibodies in the development of systemic sclerosis (SSc), particularly those not linked to well-defined autoantigens, remains largely unknown.
SSc-specific autoantibodies play a critical role in the diagnosis, differentiation and stratification of the disease.
Ongoing efforts are underway to discover novel autoantibodies using advanced techniques.
WHAT THIS STUDY ADDS
This study represents the initial identification and validation of anti-PRMT5 antibodies in two independent groups of patients with SSc.
Levels of anti-PRMT5 antibodies exhibited a correlation with the disease trajectory in SSc, serving as a predictive indicator for regression or progression in both skin and lung involvement.
Immunisation with recombinant protein PRMT5 induced SSc-like manifestations in mice, indicating a potentially pivotal role in SSc.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
Anti-PRMT5 antibodies manifested as a diagnostic and predictive marker for SSc.
The application of anti-PRMT5 antibodies may contribute to precisive disease monitoring and prognosis.
Introduction
Systemic sclerosis (SSc) is an autoimmune rheumatic disease, characteristic of autoimmunity with increased inflammatory burden, vasculopathy with extensive endothelial dysfunction and tissue fibrosis with fibroblast activation. 1–4 An abundance of autoantibodies detected in SSc and the close link with clinical outcomes indicate the potential involvement of autoantibodies and the breakdown of self-tolerance in the pathogenesis of SSc, thereby offering up important novel diagnostic and therapeutic opportunities of autoantibodies. 5 6 Nevertheless, the well-defined SSc autoantigens are ubiquitously expressed and play an essential role in physiological processes. Consequently, the strong association of specific SSc autoantibodies with clinical phenotypes raises intriguing questions regarding their roles in SSc pathogenesis: are these autoantibodies the drivers or merely incidental bystanders?
Antitopoisomerase antibodies (ATAs), anticentromere antibodies (ACAs) and anti-RNA polymerase III antibodies (ARAs), three types of antinuclear antibodies (ANAs), have been reported as the most prevalent SSc-specific antibodies. However, up to 11% of patients with SSc are negative for ANA, 7 and even as many as 17% of patients with SSc lack detectable levels of established SSc-specific antibodies. 8 This highlights the necessity to explore novel autoantibodies that are specific to SSc. There is a growing body of studies focusing on the identification of novel autoantibodies and the definition of their clinical implications, such as antiplatelet-derived growth factor receptor (PDGFR), antiangiotensin receptor type 1, antigephyrin and antieukaryotic initiation factor 2B antibodies. 9–14 Mechanistically, single-cell analysis reveals broad differences in cell cluster gene expression profiles, showing distinctions in clinical phenotypes and distinct skin score trajectories across autoantibody subgroups of diffuse cutaneous SSc (dcSSc). 15 However, further investigation is required to elucidate the precise pathomechanism of autoantibodies in SSc.
Advanced high-profile techniques, such as solid surface arrays and display technologies, 16 have been boosting the biomarker discovery. Despite considerable progress and widespread application in autoantibody discovery, 17–19 the utility of high-throughput assays, particularly protein microarrays, remains restricted by the limited amount of miniaturised test sites. This constrains the coverage of the whole proteome and the detection sensitivity and specificity for individual protein. In addition, patient heterogeneity and varied abundance of targets can limit the progress in finding specific biomarkers in serum. Therefore, a comprehensive strategy with pre-enrichment of protein targets followed by subsequent whole-proteome screening for functional properties will facilitate the investigation of serum alterations in patients. 20 21 However, this approach necessitates extensive proof-of-concept studies and wide-spectrum cohort validation.
Here, we first identified PRMT5 as a novel autoantibody target in SSc based on the automated deep efficient peptide sequencing and quantification (DEEP SEQ) mass spectrometry (MS) platform 22 for the enriched antigen-antibody complex. We then validated the prevalence of antibodies against PRMT5 in the sera of patients with SSc and demonstrated the close correlations of anti-PRMT5 antibodies with the progression or regression trajectories of skin and lung disease in SSc. Induction of SSc-like skin and lung changes in mice via immunisation with recombinant PRMT5 protein indicates anti-PRMT5 as a potential contributing antibody in the pathogenesis of SSc. Anti-PRMT5 antibodies manifested as diagnostic and predictive marker for SSc. The utilisation of anti-PRMT5 antibodies has the potential to enhance precise disease monitoring and prognosis assessment in SSc.
Global autoantibody profiling in SSc via DEEP SEQ proteomics
To uncover elusive autoantibody surrogates for SSc, we developed a global antibody profiling strategy based on the automated DEEP SEQ MS platform 22 ( figures 1A and 2A ). In brief, we constructed an antigen pool using lysates from a variety of cell lines, including human umbilical vein endothelial cells, human dermal fibroblasts, Jurkat T cells and THP-1 monocytes, as sources of antigens for subsequent proteomics profiling. The corresponding cell types have been linked to the pathogenesis of SSc. 4 13 14 Antigen-antibody complexes were then enriched by coincubating global antibodies with the antigen pool, in conjunction with immunoprecipitation (IP) using protein A/G beads. Finally, we performed in-solution on-bead trypsin digest of the complexes and labelled peptide fragments, following subsequent quantitative proteomics analysis ( figure 2A ).
- Download figure
- Open in new tab
- Download powerpoint
Overview of experimental workflow and key discoveries. (A) Peripheral blood was obtained from the patients with SSc and healthy subjects (n=3/group), and then coincubated with an antigen pool, which was prepared from a diverse array of cell lysate of endothelial cells, fibroblasts, T cells and monocytes. Then the antigen-antibody complex was enriched with protein A/G and proceeded with on-bead digestion and labelling by means of isobaric tags for relative and absolute quantitation (iTRAQ). The quantitative proteomics of these pulldowns enabled us to illuminate the presence of over-represented putative antibody targets in patients. Applying the automated deep efficient peptide sequencing and quantification (DEEP SEQ) mass spectrometry (MS) platform, 37 our immunoprecipitation (IP)-MS analysis revealed putative autoantibodies against antigen pools, which require further verification to exclude non-specific binding. (B) Peripheral blood was obtained from 90 patients with systemic sclerosis (SSc) patients, 30 patients with systemic lupus erythematosus (SLE), 8 patients with Sjögren’s syndrome (SjS) and 84 healthy donors for antibody validation. Antibodies against PRMT1, PRMT5, HK1 and CD5L in the serum were determined by ELISA. Anti-PRMT5 was identified as a specific autoantibody for SSc. The expression of PRMT5 in SSc skin was evaluated by immunofluorescence (IF) staining. (C) The levels of anti-PRMT5 antibodies were compared between healthy subjects and patients with SSc and correlated with clinical phenotypes of patients with SSc. (D) Immunisation of mice with recombinant protein PRMT5 subcutaneously at an interval of 2 weeks for a total of four times. Skin and lung tissues were collected for pathological examination and bulk RNA sequencing (RNA-seq). Skin and lung fibrosis were found in PRMT5-immunised mice, which mimicked human SSc-like changes; thus, anti-PRMT5 antibody was identified as a potential contributing antibody for SSc.
Proteomics-based discovery of autoantibodies in SSc. (A) Schematic illustration of the proteomics-based approach employed to identify autoantibodies associated with SSc. The graphic was generated using BioRender.com and complies with BioRender’s Academic License Terms. (B) Heatmap diagram displaying differentially expressed antibodies in the sera of patients with SSc compared with healthy controls. Red values indicate upregulation, while green values indicate downregulation. (C) Identification of autoantibodies by proteomics in SSc. Volcano plots illustrate iTRAQ proteomics results for enriched and purified proteins bound to antibodies in serum from patients with SSc versus healthy donors. The x-axis represents relative protein levels (mean log2 iTRAQ ratios across three replicate experiments) in patients with SSc compared with healthy donors, while the y-axis displays log10 (p values). Significantly enriched and upregulated proteins (p≤0.05; iTRAQ ratio ≥1.5) are denoted by red dots, significantly enriched and downregulated proteins (p≤0.05; iTRAQ ratio ≤−1.5) by blue dots, and all others by grey dots. The dotted lines indicate a 1.5-fold ratio (x-axis) and a p-value of 0.05 (y-axis). (D) Protein-protein interaction networks of a subset of significantly enriched and upregulated antibodies in SSc, highlighting PRMT5 as the antibody target. (E) Heatmap diagram depicting representative differentially expressed antibodies in SSc. Red values indicate upregulated proteins, while blue values indicate downregulated proteins. PRMT5 as the target is highlighted. iTRAQ, isobaric tags for relative and absolute quantitation; HC, healthy control; MS, mass spectrometry; SSc, systemic sclerosis.
In this study, sera from patients with SSc (n=3) and healthy controls (HCs) (n=3) were subjected to the detection of global antibodies ( online supplemental table S1 ). By applying the DEEP SEQ proteomics platform, we identified 4798 proteins in total (≤1% false discovery rate (FDR) at peptide level). Among these, 238 enriched proteins were significantly upregulated in SSc in contrast to HCs in IP-MS analysis targeting putative autoantibodies against antigen pools ( figure 2B,C ). Not all of the 238 identified proteins serve as targets for autoantibodies; some may exhibit non-specific binding due to the limitations of quantitative proteomics techniques. Therefore, further verification is necessary to screen for potential autoantibody candidates within this group of proteins. To confirm the validity of antigen capture process, topoisomerase I (Topo I) was detected by western blotting following IP in a patient who was positive for anti-Topo I antibody (ATA), whereas undetectable in an ATA-negative patient ( online supplemental figure S1A,B ). In addition, protein-protein interaction networks showed the emergence of protein interaction ‘hot spots’ surrounding Topo I, one of the well-defined antibody targets and frequently associated with SSc 23 ( figure 2D ). Among Topo I-associated proteomes, PRMT1, PRMT5, HK-1 and CD5L were over-represented in patients with SSc than HC ( figure 2E ). Since literature results suggested PRMT1, PRMT5, HK-1 and CD5L with promising pathophysiological relevance, 24–28 we then focused our investigations on these four proteins as potential autoantibody targets.
Supplemental material
Validation of anti-prmt5 antibody as a specific autoantibody for ssc.
To confirm the discovery results, serum levels of antibodies against PRMT1, PRMT5, HK-1 and CD5L were then measured by ELISA in a primary validation cohort, including 90 patients with SSc, 30 patients with systemic lupus erythematosus (SLE), 8 patients with Sjögren’s syndrome (SjS) and 84 sex-matched and age-matched HC ( online supplemental table S2 , figure 1B ). Serial dilutions for serum were applied to determine the optimal condition for ELISA and calculate values of area under the curve (AUC) ( online supplemental figure S2A–D ). Serum levels of antibodies against PRMT5, measured as absorbance signals at 405 nm by ELISA, were significantly higher in patients with SSc, compared with HC or the patients with SLE and SjS (SSc vs HC p<0.001; SSc vs SLE p<0.001; SSc vs SjS p=0.003) ( figure 3A ). Consistently, when calculated by the values of AUC using serial dilutions, serum levels of anti-PRMT5 antibodies also demonstrated an increase in patients with SSc relative to HC, SLE or SjS ( online supplemental figure S3A ). On the contrary, the level of anti-CD5L antibodies showed a moderate increase in patients with SSc relative to HC (p=0.019) but was comparable with the patients of SLE or SjS. No significant difference was found in serum levels of antibodies against PRMT1 or HK1 across SSc, HC, SLE and SjS ( figure 3A , online supplemental figure S3B–D ). Next, to exclude technical false positivity, non-relevant anti-ZIKV envelope DIII virus antibodies 29 were tested as undetectable in both patients with SSc and HC ( online supplemental figure S4A–C ). Furthermore, using the 99th percentile as the upper limit of normal, anti-PRMT5 antibodies were present in 31.11% of patients with SSc (28/90) and absent in HC (0/84) ( figure 3B ), with sensitivity, specificity, positive predictive value and negative predictive value of 70.24%, 97.78%, 96.72% and 77.88%, respectively. The positivity of anti-PRMT5 antibodies in SSc was greater than anti-PRMT1, HK-1 and CD5L antibodies ( figure 3C–E , online supplemental figure S3E–G ). Interestingly, anti-PRMT5 antibodies also demonstrated the ability to differentiate SSc from the patients of SLE and SjS with AUC of 0.968 and 0.988, respectively ( figure 3D, E ). Following this, we proceeded to validate the levels of anti-PRMT5 antibodies in a recently recruited, independent group of patients with SSc. Consistently, we observed significantly elevated levels of anti-PRMT5 in the sera of these patients with SSc compared with the HC ( online supplemental table S3 , online supplemental figure S5A,B ). Furthermore, we validated the elevated levels of anti-PRMT5 antibodies in sera from these patients with SSc and HC determined using microarray ( online supplemental figure S5C,D ). Overall, these results indicate that the anti-PRMT5 antibody is a specific surrogate biomarker for SSc.
Validation of anti-PRMT5 antibody as a specific autoantibody for SSc. (A) Antibodies against PRMT1, PRMT5, HK-1 and CD5L, determined by ELISA, in serum of 90 patients with systemic sclerosis (SSc), 30 patients with systemic lupus erythematosus (SLE), 8 patients with Sjögren’s syndrome (SjS) and 84 healthy controls (HCs). Data of A are presented as median±IQR, each dot representing one sample. P values were determined by Kruskal-Wallis test with Dunn’s multiple post hoc tests. P values are indicated in the figures. (B) Bar graphs demonstrating the proportion of patients positive or negative for the antibodies against PRMT5, PRMT1, HK-1 and CD5L. The positivity of serum antibody levels was determined if the values were above the 99th percentile as the upper limit of healthy donors. P values were determined by Fisher’s exact test. (C–E) Illustrations of the receiver operating characteristic (ROC) curves, plotted based on the serum levels of antibodies against PRMT5, PRMT1, HK-1 and CD5L in patients with SSc, compared with HC (C), SLE (D) and SjS (E), respectively. The values of the area under the curve (AUC) represent as indicated.
Correlations of the serum levels of anti-PRMT5 antibodies with clinical features of SSc
To define the clinical implication of anti-PRMT5 antibodies, we compared the serum antibody levels in patients with SSc with an array of clinical phenotypes ( figure 1C ). Patients exhibiting progression in skin fibrosis, as determined by a 25% increase in the modified Rodnan skin score (mRSS) compared with the previous visit within a 12-month period, demonstrated elevated serum levels of anti-PRMT5 antibodies ( figure 4A ). With respect to the relation between anti-PRMT5 antibodies and skin or lung score trajectories of the patients with SSc, we investigated if the occurrence and dynamic changes of these antibodies may fluctuate in parallel with the skin and lung changes during the disease course. Follow-up investigations revealed an elevation in anti-PRMT5 antibodies among patients exhibiting progression in mRSS and a decline in those with mRSS regression ( figure 4B ). To assess the predictive value of baseline anti-PRMT5 antibody levels for skin fibrosis progression over a prospective 12-month period, we examined patients with SSc manifesting skin fibrosis progression, which was defined as an increase in mRSS ≥25% from baseline in the follow-up visit (12 months after baseline). The patients with SSc with mRSS progression displayed a numerical elevation in baseline levels (p=0.102) of anti-PRMT5 antibody compared with the patients with SSc without skin fibrosis progression ( figure 4C ). Anti-PRMT5 antibodies also demonstrated the ability to differentiate patients with SSc with mRSS progression from the patients with SSc without mRSS progression with an AUC of 0.792 ( figure 4D ). To test the potential of anti-PRMT5 antibody as a candidate disease indicator, we took a thorough follow-up for patients with SSc, assessing skin mRSS score and examining anti-PRMT5 levels every 3 months. Remarkably, we observed a parallel change between anti-PRMT5 levels and mRSS scores ( online supplemental figure S6A,B ).
Correlations between anti-PRMT5 antibody levels and the progression of skin and lung fibrosis in patients with systemic sclerosis (SSc). (A) Comparison of anti-PRMT5 antibody levels in patients with SSc, who showed progression or no progression defined by an increase (≥ 25%) in their total mRSS score in the past 12±2 months. (B) Paired comparison analysis of anti-PRMT5 antibody levels of patients with SSc with progression or regression in total mRSS, comparing between the baseline and the follow-up mRSS score with an interval of 12±2 months. (C) Comparison of anti-PRMT5 antibody levels in patients with SSc, with and without skin fibrosis progression, was evaluated prospectively. Skin fibrosis progression was defined as an increase in the modified Rodnan skin score (mRSS) of ≥25% from baseline in the follow-up visit (12 months after baseline). Comparison data are shown as bar graphs with individual values, where each dot represents one sample, and the median and quartiles are indicated. (D) Illustrations of the receiver operating characteristic (ROC) curves, plotted based on the serum levels of antibodies against PRMT5, comparing between patients with SSc with or without mRSS progression prospectively. The values of area under the curve (AUC) represent as indicated. (E) Comparison of anti-PRMT5 antibody levels in patients with SSc-interstitial lung disease (ILD), who showed progression or no progression in their high-resolution CT (HRCT), compared with the HRCT undertaken in the past 12±2 months. (F) Paired comparison analysis of anti-PRMT5 antibody levels of patients with SSc-ILD with progression or regression in lung HRCT, comparing the baseline and the follow-up lung HRCT within an interval of 12±2 months. (G) Comparison of anti-PRMT5 antibody levels in patients with SSc developing progressive fibrosing ILD (PF-ILD) or not in the preceding 24-month follow-up. Comparison data in A, C, E and G are shown as bar graphs with individual values, each dot representing one sample, with the median shown as a continuous line and the quartiles as discontinuous lines. Data in A, C, E and G were analysed by two-sided Mann-Whitney test. Data in B and F were analysed by Wilcoxon matched-pair signed rank test. The p values are indicated in the figures, and p<0.05 was considered statistically significant.
Likewise, patients having experienced progression in lung fibrosis, determined by an increased involvement of semiquantified areas in high-resolution CT (HRCT) in the past 12 months, demonstrated a significant elevation in anti-PRMT5 antibody levels ( figure 4E ). Similarly, the trends of anti-PRMT5 antibodies exhibited a parallel change in patients with SSc with HRCT progression or regression, as compared with the follow-up HRCT score ( figure 4F ). Patients with SSc who fulfilled the criteria of progressive fibrosing interstitial lung disease (ILD) (PF-ILD) in the subsequent 24-month follow-up demonstrated significantly increased basal levels of anti-PRMT5 antibodies compared with the patients with SSc without developing PF-ILD ( figure 4G ).
Furthermore, serum anti-PRMT5 antibodies correlated positively with the levels of acute phase reactants (APRs) like erythrocyte sedimentation rate (ESR) and C reactive protein (CRP), as well as IgG and tissue inhibitor of metal protease 1 (TIMP-1) in SSc ( figure 5A ). Furthermore, patients with SSc with elevated APR levels, defined as having at least one of the following, CRP ≥6 mg/L, ESR ≥28 mm per hour or platelet count ≥330×10⁹/L, showed higher serum levels of anti-PRMT5, compared with the patients with SSc without APR elevation ( figure 5B ). According to the criteria for active disease defined in focuSSced 30 study, we found that active patients with SSc also displayed a greater abundance of anti-PRMT5 antibody ( figure 5B ). Moreover, we found positive correlations between anti-PRMT5 antibody levels and concentrations of IL-6, tumour necrosis factor alpha, IL-10 and IL-8 in patients with SSc ( online supplemental figure S6C ). Together, our data may indicate a potential link between anti-PRMT5 antibody and the inflammatory status of patients with SSc.
Correlations between anti-PRMT5 antibody levels and inflammatory and autoimmune markers in patients with systemic sclerosis (SSc). (A) Correlations of the serum levels of anti-PRMT5 antibodies with erythrocyte sedimentation rate (ESR), C reactive protein (CRP), IgG and tissue inhibitor of matrix metalloproteinase (TIMP)-1 in SSc. (B) Comparison of anti-PRMT5 antibody levels between patients with SSc with elevated acute phase reactant (APR) levels and the individuals without, stratified as having at least one of the following or not: C reactive protein ≥6 mg/L, ESR ≥28 mm per hour or platelet count ≥330×10⁹/L (left). Comparison of anti-PRMT5 antibody levels between active patients with SSc and non-active patients with SSc, stratified according to the criteria for active disease defined in focuSSced 30 study (right). (C) Comparison of the positivity of anti-PRMT5 antibodies in patients with SSc with different clinical subsets, including diffuse cutaneous SSc (dcSSc) versus limited cutaneous SSc (lcSSc), positive versus negative for anti-topoisomerase I antibody (ATA), anticentromere antibody (ACA) and anti-RNA polymerase III antibody (ARA). (D) Comparison of percentages of interstitial lung disease (ILD) between patients with SSc double positive for both ATA and anti-PRMT5 antibodies (APA) or not (left). Comparison of percentages of dcSSc versus lcSSc between patients with SSc double positive for both ATA and APA or not (right). Data in A were analysed using non-parametric Spearman correlation analysis. Data in B were analysed by two-sided Mann-Whitney test. Contingency data in C and D were analysed using Fisher’s exact test. The p values are indicated in the figures, and p<0.05 was considered statistically significant.
Patients with SSc with diffuse cutaneous involvement (dcSSc) demonstrated a relatively higher positivity for anti-PRMT5 antibody compared with the patients with SSc with limited cutaneous involvement (lcSSc) but without statistical significance (dcSSc vs lcSSc: 56.34% vs 43.66%, p=0.179, figure 5C ). Additionally, no relevance was found between anti-PRMT5 antibody and other known SSc-specific antibodies, including ATA, ACA and ARA ( figure 5C ). Interestingly, 15 out of 90 (16.67%) patients were double positive for ATA and anti-PRMT5 antibodies. Notably, within this group, 13 out of 15 (86.67%) patients manifested evidence of ILD on HRCT, a higher proportion compared with the non-double positive patients (41/75, 54.67%), with statistical significance (p=0.023, figure 5D ). Interestingly, among 18 follow-up patients, all 3 individuals who tested positive for both ATA and anti-PRMT5 antibody experienced ILD progression and fulfilled the criteria of PF-ILD within the preceding 24-month follow-up. Additionally, patients with SSc who are double positive for ATA and anti-PRMT5 antibodies also exhibited a numerical predominance for the diffuse cutaneous subset (11/15, 73.33%) compared with the limited cutaneous subset (4/15, 26.67%) but without statistical significance ( figure 5D ). No correlations were found between serum levels of anti-PRMT5 antibodies with other clinical parameters in terms of age, sex, disease duration, therapeutic backgrounds, commodity diseases, current mRSS, the presence of digital ulcer (DU), pulmonary arterial hypertension (PAH), telangiectasia or the pattern of nailfold capillaroscopy (data not shown). The data suggest that anti-PRMT5 antibodies are more closely associated with the disease trajectory observed in the skin and lungs of patients with SSc, surpassing the correlation with their current level of involvement.
Furthermore, PRMT5 was more pronounced in fibroblasts and moderately increased in endothelial cells in the dermis of patients with SSc relative to HC ( online supplemental figure S7A–C ). As the apoptosis of endothelial cells contributes to the pathogenesis of SSc as one of the initial steps, 31 32 we also observed the significantly increased cell counts of apoptotic PRMT5-positive endothelial cells in the dermis of patients with SSc ( online supplemental figure S8 ), indicating the potential underlying mechanism that PRMT5 may be exposed from apoptotic endothelial cells, triggering autoimmune response subsequently. PRMT5 was observed to be expressed in CD3 + T cells and CD68 + macrophages of skin, however, without statistical difference between patients with SSc and HC (data not shown).
Induction of skin and lung fibrosis by immunisation with PRMT5
To elucidate the contribution of anti-PRMT5 antibodies to the development of SSc, we immunised mice with recombinant protein PRMT5 ( figure 6A ). Skin fibrosis and lung fibrosis were examined histopathologically 8 weeks after initiation of PRMT5 treatment. Treatment with PRMT5/complete Freund’s adjuvant (CFA), in contrast to the treatment with vehicle (Veh)/CFA, resulted in skin fibrosis with increased dermal thickness. In addition, there was no significant difference between the mice treated with PRMT5 and Topo I ( figure 6B,C ). Similarly, ILD was observed in PRMT5/CFA-treated mice, exhibiting extensive inflammatory infiltration and diffuse fibrosis, with remarkably increased Ashcroft score than Veh/CFA-treated control mice ( figure 6D,E ). In addition, the Ashcroft score was comparable between the mice treated with PRMT5 and Topo I ( figure 6D,E ). Immunofluorescence costaining showed an increased number of α-smooth muscle actin (αSMA + ) fibroblast activation protein (FAP + ) myofibroblasts in the skin and lungs of mice treated with PRMT5/CFA, compared with the control mice treated with Veh/CFA ( figure 6F–I ).
Induction of skin and lung fibrosis in mice immunised by recombinant protein PRMT5. (A) Immunisation with recombinant protein PRMT5 or DNA topoisomerase I (Topo I), along with complete Freund’s adjuvant (CFA) four times subcutaneously with an interval of 2 weeks. Skin and lung tissue samples were collected and followed by the pathological examination (n=6 independent biological samples per group). (B) Representative H&E and Masson’s trichrome staining of the skin shown at 200-fold magnification (scale bars=100 µm). (C) Quantification of dermal thickness, which are normalised to controls. (D) Representative H&E and Masson’s trichrome staining of the lungs shown at 200-fold magnification (scale bars=100 µm). (E) Ashcroft scores were assessed and normalised to controls. (F, H) Representative immunofluorescence staining for αSMA (green) and costaining with FAP (red) in the dermis (F) or lungs (H) of mice treated with PRMT5, or Topo I, along with CFA, at 400-fold magnification (F and H; scale bars=50 µm). (G, I) Numbers of αSMA-positive fibroblasts per high power field (HPF) in the skin (G) and lungs (I) are quantified. All data are presented as median±IQR, each dot representing one sample. P values were determined by Kruskal-Wallis test with Dunn’s multiple post hoc test (C, E, G and I). P values are indicated in the figures.
To further investigate the serological antibody response, we examined the induction of anti-PRMT5 or Topo I antibodies in sera of mice immunised with recombinant PRMT5 or Topo I, emulsified in CFA, through ELISA. Our findings revealed higher levels of anti-PRMT5 antibodies in sera of mice immunised with recombinant PRMT5, while higher levels of anti-Topo I antibodies in sera of mice immunised with recombinant Topo I ( online supplemental figure S9 ). Further multiplexed immunofluorescence staining was performed by labelling with antibodies against CD45, CD3, CD68 and CD20, which have been reported as markers for pronounced infiltrating immune cell types in SSc. 1 3 We found a remarkable increase in immune cell infiltration in the skin of PRMT5 immunised mice, including T cells, macrophages and B cells ( online supplemental figure S10A,B ). Immune infiltration dominant by T cells and macrophages was also found in the lungs of the mice immunised by PRMT5 plus CFA, compared with the Veh /CFA-treated control mice ( online supplemental figure S10C,D ).
To further explore the impact of anti-PRMT5 antibodies on mice and unravel underlying mechanism, we conducted RNA-seq analysis on skin and lung tissues obtained from the mice immunised with PRMT5/CFA or Veh/CFA. Applying a threshold of p<0.05 and |log2 fold change (FC)| ≥1, we identified a total of 4205 and 1169 differentially expressed genes (DEGs) in the skin and lungs, respectively (in the skin, 2681 upregulated DEGs and 1524 downregulated DEGs; in the lungs: 640 upregulated DEGs and 529 downregulated DEGs), comparing between PRMT5/CFA-treated mice versus Veh/CFA-treated control mice ( online supplemental figure S11A,B,E,F ). The gene sets encompass numerous genes previously implicated in the pathogenesis of SSc, such as Acta2 , Col1a1 , Col3a1 , Smad3 , Ctgf , Msr1 , Cd4 , Cd8a and Cd68 , supporting the potential of anti-PRMT5 in induction of SSc-like skin changes. Furthermore, the upregulated gene set identified in the lungs of PRMT5/CFA immunised mice included the overlapping DEGs observed in the skin, such as Smad3 , Msr1 and Cd8a , along with other fibrosis-associated genes like Cd163 , Shh , Edn1 and Fgf12 ( online supplemental figure S11E,F ).
To elucidate the functional associations of the identified gene signatures, we performed a gene enrichment analysis for Gene Ontology (GO). This analysis revealed the enrichment of genes involved in immune response and extracellular matrix organisation as key biological processes in the skin of PRMT5/CFA-immunised mice, suggestive of the activation of the immune system and fibrosis process that mirrors characteristics observed in patients with SSc ( online supplemental figure S11C ). Consistent with this notion, we observed the upregulation of the signalling pathways previously associated with SSc, 3 33 such as interleukin (IL)-6, IL-4, IL-17, IL-1β, toll-like receptor, vascular endothelial growth factor (VEGF) signalling, JAK-STAT signalling as well as pathways involved in wound healing and extracellular matrix organisation ( online supplemental figure S11C ). Further analysis using Reactome database demonstrated changes in VEGF or PDGF-mediated signalling, antigen presentation, extracellular matrix organisation, assembly of collagen fibrils as well as cell surface interaction at the vascular wall ( online supplemental figure S11D ).
In addition, systemic analysis using GO and Reactome databases demonstrated several terms relevant to SSc in the lungs of PRMT5/CFA-immunised mice, including ‘response to IL-13’, ‘wound healing involved in inflammatory response’, ‘response to hypoxia’, ‘antigen receptor-mediated signalling pathway’, ‘classical antibody-mediated complement activation’ and ‘immunoregulatory interactions between a lymphoid and a non-lymphoid cell’ ( online supplemental figure S11G,H ).
We then delved into signalling pathways governed by DEGs via further Ingenuity Pathway Analysis (IPA). Alongside the activation of numerous fundamental immune and inflammatory responses, as well as cytokine signalling in skin tissues from the mice immunised with PRMT5 ( online supplemental figure S12A ), our analysis pinpointed the upregulation of a cascade of fibrosis-associated signalling pathways integral to the pathogenesis process, 4 33 including STAT3, PDGF and G-proteincoupled receptor ( online supplemental figure S12 ). Signalling pathways associated with tissue and matrix regulation have been over-represented in the skin tissues of PRMT5 immunised mice, such as ‘wound healing signalling pathway’, ‘extracellular matrix organisation’ and ‘collagen biosynthesis and modifying enzymes’. Conversely, the suppression signal like ‘inhibition of matrix metalloproteases’ was significantly inhibited ( online supplemental figure S12A ). This comprehensive understanding is summarised through a graphical network encompassing canonical pathways, upstream regulators and biological functions ( online supplemental figure S12B ).
Therefore, immunisation with PRMT5 provokes SSc-mimicking inflammation and fibrosis in the skin and lungs in vivo. These findings may indicate PRMT5 as a potential contributing antibody target in SSc, likely participating in the processes of autoimmune response and myofibroblast activation.
Our study here is the first to identify PRMT5 as a novel autoantibody target of the autoimmune response in patients with SSc based on large-scale proteomics with automated DEEP SEQ. 22 Anti-PRMT5 antibodies are present in 31.11% of patients with SSc and absent in HCs. We found the dynamic changes of anti-PRMT5 antibodies in parallel with the progression or regression of skin fibrosis and ILD of SSc; monitoring the levels of anti-PRMT5 antibodies may therefore enable early detection and the initiation of early intervention for the patients with SSc with a higher risk of mRSS progression and development to PF-ILD during follow-up. Histopathological evaluations further revealed an overexpression of PRMT5, predominantly localised within fibroblasts and endothelial cells, among patients with SSc. Furthermore, mice immunised with PRMT5 showed marked tissue fibrosis coupled with immune cell infiltration within both dermal and pulmonary tissues, as well as serological antibody response, mirroring the pathognomonic changes typically associated with human SSc. RNA-seq demonstrated immunisation with PRMT5-induced multiple profibrotic and proinflammatory transcriptional networks in mice. Collectively, these data thus indicate the potential role of anti-PRMT5 as a contributing autoantibody in the development of SSc.
The first step of the current study is a screening for autoantigens with an exploratory strategy using high-throughput proteomics. Lysates of the cell lines were used as a source of putative autoantigens. The antibody-antigen complex was enriched via IP and analysed with large-scale proteomics based on automated DEEP SEQ MS as we established previously. 22 In contrast to the DNA sequencing platforms, the protein-level array poses challenges attributed to the wide dynamic span in protein expressions and vast diversity in post-translational modifications, coupled with the lack of an amplification strategy analogous to PCR, limiting genome-wide protein characterisation, particularly for signal transduction and other key regulatory factors that are often present in low abundance. To address this issue, we employed an established genome-scale proteome quantification by DEEP SEQ MS, which is based on simple detergent lysis and single-enzyme digest, extreme, orthogonal separation of peptides and true nanoflow liquid chromatography (LC)-MS/MS, significantly increasing the scale of proteome coverage. 22 In fact, omitting protein crosslinking may avoid the complexities associated with diverse protein conformations. Subsequent studies in patient cohort demonstrated that the autoantibodies against PRMT5 can be detected in SSc with high diagnostic performance. In this case, these results support the feasibility of this coordinated workflow combining thorough screen and functional validation for the identification of optimal diagnostic and therapeutic antibody candidates in general.
The presence of autoantibodies in patients does not mean that the autoantibodies can mediate the clinical manifestations. Therefore, the challenge is to clarify the role of autoreactivity in a clinical scenario and determine whether autoreactivity is crucial or merely incidental. The validity of anti-PRMT5 antibodies rather than anti-PRMT1, HK-1 or CD5L antibodies with higher specificity and sensitivity for SSc leads to the next in-depth investigation for anti-PRMT5 in SSc. Besides, anti-PRMT5 antibodies demonstrated promising predictive value for the progression of skin and lung disease in SSc. Therefore, the role of autoimmunity of anti-PRMT5 was then assessed in an immunisation model, which has been extensively used for model establishment, such as experimental allergic encephalomyelitis, 34 collagen-induced arthritis, 35 glucose-6-phosphate isomerase-induced arthritis 36 as well as Topo I immunised SSc. 23 Indeed, induction of tissue fibrosis and immune infiltration in mice immunised by recombinant protein PRMT5 illustrated that the antibody response to PRMT5 was likely to result in SSc-like manifestations. As anticipated, we observed several upregulated DEGs in he skin related to T cell, B cell and macrophage response, as well as fibroblast activation. Through GO and Reactome analysis, we revealed characteristic changes in both the skin and lungs of PRMT5-immunised mice, clearly distinguishing them from controls. These changes included several key functional categories: (1) we observed various biological processes in PRMT5-immunised mice related to autoimmune responses, with particular emphasis on processes linked to antigen presentation and immune cell activation; (2) we showed the increased production of profibrotic cytokines, including IL-4, IL-13, IL-6 and IL-1β; (3) we found multiple biological processes related to fibroblast activation and fibrotic tissue remodelling, including extracellular matrix organisation, biosynthesis and assembly; and (4) we observed the activity of several key profibrotic signalling, such as VEGF, PDGF, JAK-STAT and Toll-like receptor-mediated signalling. Further comprehensive analysis encompassing canonical pathways, upstream regulators and biological functions was also conducted by IPA. Thus, our unbiased RNA-seq analysis highlights immune response and tissue remodelling as a characteristic feature across the skin and lungs in the mice immunised with PRMT5, mirroring aspects of human SSc pathology.
The overexpression of PRMT5 in the fibroblasts and endothelial cells of SSc has provided the rationale that PRMT5 might be related to the fibroblast activation and endothelial dysfunction, which have been revealed as crucial in the pathogenesis of SSc. 2 4 PRMTs have been shown to play critical roles in disease through methylation of arginine residues on histone or non-histone proteins. Of note, circulating monomethyl arginine and asymmetrically dimethylated arginine can inhibit the function of nitric oxide (NO) synthase, which generates NO. Interestingly, attenuated NO bioavailability results in a milieu of inflammation and oxidative stress in SSc, leading to vasculopathy and subsequent fibrosis and reshaping of NO metabolism has been proven to be an effective treatment of SSc-associated vasculopathy, especially for DU and PAH. 37 38 PRMT5 was identified as a symmetrical dimethyltransferase ubiquitously expressed in the kidneys, skin, lungs and other tissues. 39 PRMT5 inhibitors have demonstrated efficiency in treating mouse models of acute graft-versus-host disease, as elucidated by prolonged survival and ameliorated disease severity, along with decreased T cell proliferation and cytokine production. 40 PRMT5 has been reported to regulate T cells through various pathways, including promoting retinoid-related orphan receptor (ROR)-γt activity and adjusting the Klf2-S1pr1 pathway. 41 42 Arginine methylation mediated by PRMTs has emerged as a critical mechanism implicated in fibrosis. 43–47 Notably, fibroblast-specific deletion of PRMT5 significantly reduced pressure overload-induced cardiac fibrosis. PRMT5 has been shown to regulate transforming growth factor beta (TGF-β)/Smad3-dependent fibrotic gene transcription, potentially through histone methylation crosstalk, and plays a critical role in cardiac fibrosis and dysfunction. 45 Similarly, the contribution of PRMT5 to fibrosis has been confirmed in an Adriamycin-induced cardiac fibrosis model. 46 These findings suggest that PRMT5 may serve as a critical mediator in regulating TGF-β-stimulated fibroblast activation and tissue fibrosis. Since protein methylation is a targetable modification and advanced drug development of PRMT5 inhibitors has been achieved, the therapeutic potential of targeting PRMT5 appears promising.
In sum, this study has identified anti-PRMT5 antibodies as a novel biomarker for SSc. The current data suggest the potential underlying mechanism driven by PRMT5 as a target of autoimmunity and consequently resulting fibrosis in SSc. However, the exact role of anti-PRMT5 in SSc needs further elucidation.
A detailed description of all materials and methods is provided in online supplemental material .
All human studies were approved by the ethical committee of the Medical Faculty of Fudan University. All patients and controls signed an informed consent form approved by the local institutional review board.
All animal experiments were carried out in strict accordance with international and local guidelines for animal care and use. Mice were maintained under pathogen-free conditions, with a standard diet, water ad libitum and 12 hour light/12 hour dark cycle. Mice were 6-week old at the start of experiments, and up to six mice were housed in one cage.
Statistical measures, including the number of samples, descriptive statistics (median and IQR) and significance, are reported in the figures and figure legends. P<0.05 was considered statistically significant.
Ethics statements
Patient consent for publication.
Not applicable.
Ethics approval
The protocol of mouse experiment was approved by the Fudan University, Shanghai, PRC (2021JS HSYY-052; 2022JS HSYY-054; 2023-HSYY-179JZS). This study involves human participants and was approved by Huashan Hospital Institutional Review Board. Participants gave informed consent to participate in the study before taking part.
Acknowledgments
We would particularly like to acknowledge my colleagues at the Division of Rheumatology of Huashan Hospital for their wonderful collaboration and patient support. We thank AbCode Co, Ltd, for the support of microarray assay.
- Gabrielli A ,
- Avvedimento EV ,
- Denton CP ,
- Chakraborty D ,
- Distler JHW
- Distler JHW ,
- Györfi A-H ,
- Ramanujam M , et al
- Patterson K , et al
- Pisetsky DS
- Nihtyanova SI ,
- Chen C , et al
- Wang X , et al
- McMahan ZH ,
- Kulkarni S ,
- Andrade F , et al
- Favoino E ,
- Cipriani P ,
- Liakouli V , et al
- Baroni SS ,
- Santillo M ,
- Bevilacqua F , et al
- Riemekasten G ,
- Pauling JD ,
- Salazar G ,
- Lu H , et al
- Clark KEN ,
- Attah M , et al
- Carlton LH ,
- McGregor R ,
- Moreland NJ
- Ho J , et al
- Sjöberg R ,
- Mattsson C ,
- Andersson E , et al
- Jaycox JR ,
- Vulsteke J-B ,
- Dubucquoi S , et al
- Storey AJ ,
- Hassen SI , et al
- Ficarro SB , et al
- Yoshizaki A ,
- Ogawa A , et al
- Sengupta S ,
- Kennemer A ,
- Patrick K , et al
- Edell C , et al
- Huang W , et al
- Iannaccone A ,
- Hollingsworth TJ ,
- Koirala D , et al
- Gaublomme J , et al
- Du L , et al
- Furst DE , et al
- Mostmans Y ,
- Giddelo C , et al
- Maehara T ,
- Perugino CA , et al
- Kerlero de Rosbo N ,
- Meehan GR ,
- Al Khabouri S , et al
- Krämer A , et al
- Girgis RE ,
- Gugnani MK ,
- Abrams J , et al
- Aubourg F , et al
- Fagerberg L ,
- Hallström BM , et al
- Snyder KJ ,
- Zitzer NC ,
- Gao Y , et al
- Kim H-Y , et al
- Zhou B , et al
- Zakrzewicz D ,
- Zakrzewicz A ,
- Didiasova M , et al
- Katanasaka Y ,
- Murata N , et al
- Yu S-Z , et al
- Liu F , et al
Supplementary materials
Supplementary data.
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1
- Data supplement 2
- Data supplement 3
- Data supplement 4
- Data supplement 5
- Data supplement 6
- Data supplement 7
- Data supplement 8
- Data supplement 9
- Data supplement 10
- Data supplement 11
- Data supplement 12
- Data supplement 13
Handling editor Josef S Smolen
ML, LW, XT, KW and XZ contributed equally.
Correction notice This article has been corrected since it published Online First. The legend for figure 3 has been corrected.
Contributors Overall content as the guarantors: MRL, TLY, FZ. Conceptualisation: ML, TY, FZ. Methodology: LBW, XLT, KW, QL, XYZ, TLY, FZ, XYZ. Investigation: MRL, LBW, XLT, KW, QL, XYZ, TLY, FZ. Visualisation: MRL, LBW, XLT, KW, XYZ. Funding acquisition: MRL, HJZ, TLY, FZ. Project administration: MRL, TLY, FZ. Supervision: MRL, TLY, FZ. Writing—original draft: MRL, FZ. Writing—review and editing: MRL, LBW, XLT, KW, XYZ, QL, LLH, WJY, HHY, XXZ, YX, WGW, YLW, XJC, LWL, JCW, HJZ, TLY, FZ.
Funding This study was also supported by the National Natural Science Foundation of China (NSFC) (82371818), Joint Sino-German research project from NSFC and Deutsche Forschungsgemeinschaft (German Research Foundation) (82161138022), NSFC (82030003) and Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences (2019-I2M-5-066).
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Read the full text or download the PDF:
Thank you for visiting nature.com. You are using a browser version with limited support for CSS. To obtain the best experience, we recommend you use a more up to date browser (or turn off compatibility mode in Internet Explorer). In the meantime, to ensure continued support, we are displaying the site without styles and JavaScript.
- View all journals
Antigen presentation articles from across Nature Portfolio
Antigen presentation is the process by which protein antigen is presented to lymphocytes in the form of short peptide fragments. These are associated with antigen-presenting molecules such as MHC class I or MHC class II on the surface of antigen-presenting cells (APCs).
Latest Research and Reviews
Structures of human γδ t cell receptor–cd3 complex.
- Bangdong Huang

Autophagy degrades immunogenic endogenous retroelements induced by 5-azacytidine in acute myeloid leukemia
- Nandita Noronha
- Chantal Durette
- Gregory Ehx

FcRn regulates antigen presentation in dendritic cells downstream of DEC205-targeted vaccines
- Christophe Macri
- Matthew Paxman
- Justine D. Mintern

Personalized neoantigen vaccine and pembrolizumab in advanced hepatocellular carcinoma: a phase 1/2 trial
Treatment of patients with advanced hepatocellular carcinoma with a personalized DNA vaccine in combination with anti-PD-1 therapy was safe and led to encouraging clinical efficacy, with immunological analyses confirming the induction of tumor antigen-specific T cell responses.
- Mark Yarchoan
- Edward J. Gane
- Niranjan Y. Sardesai

MARS an improved de novo peptide candidate selection method for non-canonical antigen target discovery in cancer
Detection of neoepitopes from tumours is time consuming and requires the integration of genomic and/or RNA sequencing expression data. Here, the authors propose a machine learning method to enable direct identification of additional, tumour-specific sequences using mass spectrometry through integration of de novo peptide sequencing scores, MHC class I binding prediction, and peptide retention time prediction.
- Hanqing Liao
- Carolina Barra
- Nicola Ternette

A diversity of novel type-2 innate lymphoid cell subpopulations revealed during tumour expansion
A study reveals that unique subtypes of innate lymphoid cells (ILC2s) contribute to diverse functions 53 such as tissue remodeling, immune responses, and anti-tumor CTL responses, challenging current 54 understandings of ILC2 biology.
- Clara Wenjing Xia
- Iryna Saranchova
- Wilfred A. Jefferies
News and Comment

Primary sclerosing cholangitis may drive colon cancer through antigen-driven inflammation
Primary sclerosing cholangitis dramatically increases the risk of colon cancer in patients with inflammatory bowel disease. Our study associates a newly discovered antigen-driven adaptive immune signature with the development of colorectal cancer in patients with primary sclerosing cholangitis and might help explain the high incidence of colon cancer in those patients.
Thymus mimetic cells
- Ioana Visan

CD137 ligand interacts with CD32a to trigger reverse CD137 ligand signaling
- Herbert Schwarz
Bat adaptive immunity
- Zoltan Fehervari

Helping tumour antigens to the surface
Using high-resolution proteomics to analyse clinical samples from patients with advanced melanoma, Harel et al. show that the metabolic state of melanoma is associated with changes in antigen presentation-related proteins and thereby with response to immunotherapies.
- Ulrike Harjes
cDC1 cross-priming
- Laurie A. Dempsey
Quick links
- Explore articles by subject
- Guide to authors
- Editorial policies

IMAGES
VIDEO
COMMENTS
Antigen presentation is a vital immune process that is essential for T cell immune response triggering. Because T cells recognize only fragmented antigens displayed on cell surfaces, antigen processing must occur before the antigen fragment can be recognized by a T-cell receptor. Specifically, the fragment, bound to the major histocompatibility ...
Abstract. Antigen processing and presentation are the cornerstones of adaptive immunity. B cells cannot generate high-affinity antibodies without T cell help. CD4 + T cells, which provide such ...
Learn how antigens are processed and presented by MHC class I and class II molecules to activate T cells. Explore the pathways, mechanisms and polymorphisms of antigen presentation with examples and diagrams.
MHC complexes. Antigen processing and presentation enable the adaptive immune system to survey the host cell proteome and detect pathogens and mutations 36,37.MHC I and MHC II are the two ...
Next, we review the evidence for defects in antigen presentation observed in human cancers, with a focus on understanding to what extent tumours abrogate, modulate or alter HLA-I presentation, and ...
Abstract. Antigen processing and presentation are the cornerstones of adaptive immunity. B cells cannot generate high-affinity antibodies without T cell help. CD4 + T cells, which provide such help, use antigen-specific receptors that recognize major histocompatibility complex (MHC) molecules in complex with peptide cargo.
Cross-presentation is the transferring of extracellular antigens like bacteria, some tumor antigens, and antigens in cells infected by viruses into the class I pathway for stimulation of CD8 + cytotoxic T cells (CTL). Only certain "professional" antigen-presenting cells (APCs) like dendritic cells can do this - use the class I as well as the ...
Learn about antigen presentation, a process that allows T cells to recognize antigenic epitopes displayed on the surface of antigen-presenting cells. Explore various aspects of antigen presentation, such as MHC class I and II molecules, proteasomes, transporters, and immune evasion strategies.
The three of us were fortunate to have lived through this era as young immunologists, awestruck by each new paper that modified our view of antigen presentation. In this special issue of Immunogenetics, we take you from Jan Klein's 1986 history chapter entitled "The Story" to the modern-day perspectives of antigen processing and presentation.
Learn how antigens are processed and presented by APCs on MHC molecules for T cell recognition. Explore the roles of MHC classes, TCRs, co-receptors, and autoimmune diseases.
Antigen Processing and Presentation. In Primer to the Immune Response (Second Edition), 2014. A Overview of Antigen Processing and Presentation. Antigen processing is the complex process by which antigens are produced from macromolecules. Most often, antigen processing refers to the generation of antigenic peptides from proteins.
Antigen presentation is a process in the body's immune system by which macrophages, dendritic cells and other cell types capture antigens, then present them to naive T-cells. The basis of adaptive immunity lies in the capacity of immune cells to distinguish between the body's own cells and infectious pathogens. The host's cells express ...
Antigen Presentation to T Lymphocytes. In an adaptive immune response, antigen is recognized by two distinct sets of highly variable receptor molecules—the immunoglobulins that serve as antigen receptors on B cells and the antigen-specific receptors of T cells. As we saw in Chapter 3, T cells recognize only antigens that are displayed on cell ...
Antigen presentation is the mechanism by which the antigenic environment is sampled and information imparted to the effector arms of the adaptive immune system, B and T lymphocytes. Depending on the precise context, antigen presentation can result in either activation or tolerization of lymphocytes, respective examples being the response to a ...
Learn about the essentials of antigen processing and presentation, the pathways that enable glycoproteins encoded by the MHC class I and class II molecules to be loaded with their proper ligands and presented to T cells. This Review covers the structure, function and evolution of MHC molecules, the sources and types of antigens, and the roles of TAP and tapasin in antigen presentation.
Abstract | Antigen processing and presentation are the cornerstones of adaptive immunity. B cells cannot generate high- affinity antibodies without T cell help. CD4 T cells, which provide. such ...
Professional antigen presenting cells (APCs) are immune cells that specialize in presenting an antigen to a T-cell. The main types of professional APCs are dendritic cells (DC), macrophages, and B cells. A professional APC takes up an antigen, processes it, and returns part of it to its surface, along with a class II major histocompatibility ...
Antigen Presentation with MHC I Molecules. MHC I molecules, found on all normal, healthy, nucleated cells, signal to the immune system that the cell is a normal "self" cell. In a healthy cell, proteins normally found in the cytoplasm are degraded by proteasomes (enzyme complexes responsible for degradation and processing of proteins) and ...
The mechanisms of antigen processing and presentation play a crucial role in the recognition and targeting of cancer cells by the immune system. Cancer cells can evade the immune system by downregulating or losing the expression of the proteins recognized by the immune cells as antigens, creating an immunosuppressive microenvironment, and altering their ability to process and present antigens ...
Certain APCs can phagocytose and process extracellular antigens with MHC class I molecules and then present these antigen-MHC I complexes to CD8+ T cells. This process is known as cross-presentation, or cross-priming. Cross-priming allows CD8+ T cells (which are normally activated by intracellular antigens) to be primed by extracellular ...
Tumor antigen processing and presentation is a key correlate of ICB efficacy in patients with SCLC. As antigen presentation machinery is frequently epigenetically suppressed in SCLC, this study defines a targetable mechanism by which we might improve clinical benefit of ICB for patients with SCLC.
Antigen processing and presentation is the mechanism by which whole antigens are degraded and loaded onto MHC molecules for display on the cell surface for recognition by T cells. Both macrophages ...
On presentation, the patient was febrile with a temperature of 38.3° Celsius, blood pressure of 134/99 mmHg, pulse rate of 152 beats/min, respiratory rate of 20 breaths/min, and oxygen saturation of 99% in room air. ... double-stranded DNA antibody (dsDNA), extractable nuclear antigens, anti-smith, and SMRNP antibodies with low complement ...
The enriched antigen-antibody complex was proceeded with mass spectrometry (MS)-based quantitative proteomics and over-represented by bioinformatics analysis. ... with particular emphasis on processes linked to antigen presentation and immune cell activation; (2) we showed the increased production of profibrotic cytokines, including IL-4, IL-13 ...
The field of antigen processing and presentation has continued to advance since the publication of a focus issue on the topic in Nature Immunology in July 2004. Progress has been made on many ...
Rakuten Medical's Poster Presentation. ... (EGFR), a cancer antigen expressed in multiple types of solid tumors, including head and neck, breast, lung, colorectal, prostate and pancreatic cancers ...
At the NXC-201 ASGCT 2024 late-breaking oral presentation, data were presented from 13 relapsed/refractory AL amyloidosis patients (including 3 new patients) in the ongoing Phase 1b/2a NEXICART-1 ...
Antigen presentation is the process by which protein antigen is presented to lymphocytes in the form of short peptide fragments. These are associated with antigen-presenting molecules such as MHC ...